

TO THE EDITOR:
Several studies showed that skeletal muscle myosin (SkM) and cardiac myosin (CM) can support prothrombinase complex (factor [F]Xa-FVa-Ca++) activity that generates thrombin and also support downregulation of thrombin generation by activated protein C-protein S proteolysis of FVa.1-5 Those studies included data indicating an essential role for the myosin protein component of SkM and CM preparations for their prothrombinase activity. First, antimyosin antibodies (Abs) inhibit the ability of SkM and CM to support prothrombinase. Furthermore, immobilized anti-myosin Abs remove prothrombinase supporting activity from SkM and CM preparations.1,3 Second, studies showed that the SkM heavy chain peptide HC815-837 inhibits SkM-enhanced prothrombinase and directly binds FXa, which strongly implies that it inhibits protein-protein interactions involving direct binding of FXa to SkM.2 Notably, peptide HC815-837 does not inhibit phospholipid (PL)-enhanced prothrombinase, emphasizing its direct effect on SkM-FXa interactions. Third, as noted below, FXa lacking its gamma-carboxyglutamic acid (Gla) domain (DG-FXa) retains 30% prothrombinase activity, consistent with its direct binding to SkM and CM.1,3
Recently, an important new insight into additional requirements for the prothrombinase activity of SkM and CM comes from the discovery that annexin V, lactadherin, and phospholipase A2 neutralized the prothrombinase activity of SkM and CM.6 However, that report tendentiously concluded that the prothrombinase activity of these myosin preparations was caused entirely by contaminants specifically identified as phosphatidylserine (PS)-containing PL vesicles that copurify with myosin protein. That report also suggested that the procoagulant activity of SkM and CM preparations in plasma is caused by tissue factor (TF) contaminants.6 We evaluated all of the elements of that report and, after integrating all reliable data, we provide here an updated model1 for the procoagulant activity of SkM-PL and CM-PL that involves myosin-associated anionic PLs.
First, studies confirm that annexin V potently inhibits the prothrombinase activity of SkM and CM (Figure 1A). Next, the anti-PS monoclonal Ab (mAb) 11.31 (mAb PGN632), whose reported Kd for PS is 0.2 to 1.0 nM,7-9 was used to assess whether contaminating PS could explain the prothrombinase-enhancing activity of SkM and CM preparations. Low concentrations (<1 nM) of this anti-PS mAb potently inhibited prothrombinase enhancement by phosphatidylcholine (PC)/PS (80%/20%) large unilamellar vesicles (LUVs) but not by SkM or CM (Figure 1B). However, very high levels (>50 nM) of this anti-PS mAb inhibited the prothrombinase activity of SkM and CM (Figure 1B), and the 50% inhibitory concentration (IC50) values were 60 to 100 nM, which differed by >100-fold from the IC50 value for extruded PC/PS LUVs. Because the curvature of PL surfaces can alter the prothrombinase activity of PS-containing vesicles,10 and since nothing is known about the localization of PS that is potentially associated with SkM and CM, studies tested the ability of the anti-PS mAb to inhibit the procoagulant activity of PS in large multilamellar vesicles (LMVs) and small unilamellar vesicles (SUVs). For LMVs and SUVs, the observed IC50 values of 0.4 to 0.9 nM were similar to those for LUVs (Figure 1C), implying that the prothrombinase blocking activity of mAb 11.31 is relatively independent of surface curvature. Thus, the inability of anti-PS mAb 11.31 to potently inhibit SkM or CM is not likely related to any subtle curvature feature of PS-containing PL vesicles associated with myosins.
![Effects of annexin V, anti-PS mAb, anti-TF mAb, and anti-FVII mAb on the procoagulant activities of SkM, CM, and PC/PS vesicles. (A-B) Annexin V (A) and anti-PS mAb 11.31 (B) were tested for inhibition of prothrombinase activity supported by SkM, CM, or PC/PS LUVs (PC/PS, 80%/20% w/w). Each inhibitor was incubated with FVa (2 nM, final) and FXa (0.24 nM, final) in Tris buffered saline (TBS) containing 0.5% bovine serum albumin (TBSA) and 5 mM CaCl2 in the presence of SkM (2.5 nM final), CM (2.5 nM final), or PC/PS vesicles (10 nM final). Addition of prothrombin (0.75 µM final) at room temperature initiated thrombin generation for 5 minutes that was quenched by EDTA (10 mM final). Thrombin activity was quantified by hydrolysis of the thrombin substrate (H-D-Phe-Pip-Arg-pNA). (C) Anti-PS mAb 11.31 was tested for inhibition of the prothrombinase activity supported by different sizes of PC/PS (80%/20% w/w) vesicles, as previously described. Large LMVs were prepared in TBS by vortexing a dried lipid suspension until all lipid was suspended.10 SUVs were prepared by sonication as described.1-3 LUVs were prepared by extruding LMV suspensions 20 times back and forth (total of 10 passes through the membrane) through a 0.1-μm polycarbonate membrane with a Mini-Extruder (Avanti Polar Lipids).10 (D) The effects of SkM, CM, or PC/PS LUV (PC/PS, 80%/20% w/w) on the initial rate of prothrombin (solid circles) or DG-prothrombin (open circles) activation by FXa and FVa were determined. Various concentrations of SkM, CM, or PC/PS (80%/20%) LUVs were incubated with FVa (2.4 nM final) and FXa (0.2 nM final) in TBSA plus 5 mM CaCl2. Thrombin generation was initiated by the addition of prothrombin or DG-prothrombin (0.75 μM final) and, after 10 minutes, was quenched by adding EDTA (10 mM final). The rate of thrombin formation was quantified by hydrolysis of thrombin fluorogenetic substrate. (E) FX activation assays were used to determine TF-like activity in SkM and CM preparations. Various concentrations of human or rabbit TF (Innovin, Dade or Pacific Hemostasis Thromboplastin-DS), SkM, or CM were mixed with FVIIa (0.5 nM final) in TBSA 5 mM CaCl2, and the addition of FX (0.25 μM final) initiated the reaction. After 10 minutes at room temperature, the addition of EDTA (10 mM final) quenched the reaction, and the rate of FXa formation was quantified by hydrolysis of an FXa chromogenic substrate (BIOPHEN CS11 [22]; Aniara, West Chester, OH). (F) The inhibitory effect of anti-FVII mAb on FX activation by FVIIa with human or rabbit TF was determined. Anti-FVII mAb 3G1215 (20 µg/mL final) was preincubated with FVIIa for 10 minutes at room temperature. Then TF (1.5 pM) with FX (final 250 nM) plus 5 mM CaCl2 was added and, after 10 minutes the reaction was quenched with EDTA (10 mM final). The rate of FXa formation was quantified by FXa activity (BIOPHEN CS11 [22]). (G) The effects of anti-FVII mAb or anti-TF mAb 3A5 on FX activation by FVIIa in the presence of rabbit TF, SkM, or CM was determined. Preincubation of anti-FVII mAb 3G12 (20 µg/mL, final) with FVIIa (0.5 nM final) for 10 minutes at room temperature was followed by the addition of FX (250 nM final) with rabbit TF (1.5 pM) or SkM (300 nM) or CM (50 nM) to start the reaction. After 10 minutes, the reaction was quenched by adding EDTA (10 mM final) and FXa activity (BIOPHEN CS11 [22]) was determined. For the anti-TF mAb experiment, preincubation of anti-TF mAb (RB TF 7-3A5)16 (20 µg/mL) with rabbit TF (1.5 pM) or SkM (300 nM) or CM (50 nM) for 10 minutes at room temperature was followed by the addition of FVIIa (0.5 nM final) and then FX (final 250 nM) in TBSA plus 5 mM CaCl2 to start the reaction. After 10 minutes, the reaction was quenched by adding EDTA (10 mM final) and FXa activity (BIOPHEN CS11 [22]) was determined. The 100% value was defined as FXa generated in the absence of added mAb. (H-J) The effects of anti-TF 3A5 mAb and anti-FVII 3G12 mAb on the procoagulant activity of SkM and CM in modified APTT plasma clotting assays that were previously described for the procoagulant activity of myosins.4 After preincubating SkM (H-I) or CM (J) or vehicle control buffer aliquots with anti-TF mAb (50 µg/mL) (H) or anti-FVII mAb (20 µg/mL) (I-J) for 10 minutes, the procoagulant activity was determined in the APTT assays. Each study included clotting assays for SkM and CM performed with (dashed lines) or without (solid lines) the addition of PC/PS (80%/20%) LUVs (1.0 µM final). For all studies in Figure 1 using SkM and CM, as for our published studies,1-5,17 the SkM preparation was dissolved and then dialyzed in Tris buffer (pH 7.4) containing 0.6 M NaCl at 4°C. After dialysis, particles causing turbidity were removed by high speed centrifugation (21 130g for 1 minute), which removed visible aggregates.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/13/10.1182_blood.2020008580/2/m_bloodbld2020008580f1.png?Expires=1769089025&Signature=JXkClGUPToVjzERkRR8~J4vrYL-yEe2VNHTs8NI4Zfncm34sVDL1ZgrsLVGP5jbw3Kj23aACe5AzRe1OimQBiHreaPy7H4bHhB3OPIW4m9cNpeBPBGC80FHyGEyBMOsoOddP~ponizSAM0TtgJe4whnBuQAvJLTJfQyC65RZEJDK0mgv5~WQAJMuGVoZ39LMsdn4GrUAHxQo6eVqm0k9HJZQeQsAl--YWWUncsmRxDCbJBOATMtAGh4Hl8Ntat6Fj2uMP3Jxtwa0-UXPH~00O-slKk21yRt~4d5h28a5oHYXVHkiT2pVR0DTYo0MjOvHeut813dhM2f7DAbf1e~hhw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Effects of annexin V, anti-PS mAb, anti-TF mAb, and anti-FVII mAb on the procoagulant activities of SkM, CM, and PC/PS vesicles. (A-B) Annexin V (A) and anti-PS mAb 11.31 (B) were tested for inhibition of prothrombinase activity supported by SkM, CM, or PC/PS LUVs (PC/PS, 80%/20% w/w). Each inhibitor was incubated with FVa (2 nM, final) and FXa (0.24 nM, final) in Tris buffered saline (TBS) containing 0.5% bovine serum albumin (TBSA) and 5 mM CaCl2 in the presence of SkM (2.5 nM final), CM (2.5 nM final), or PC/PS vesicles (10 nM final). Addition of prothrombin (0.75 µM final) at room temperature initiated thrombin generation for 5 minutes that was quenched by EDTA (10 mM final). Thrombin activity was quantified by hydrolysis of the thrombin substrate (H-D-Phe-Pip-Arg-pNA). (C) Anti-PS mAb 11.31 was tested for inhibition of the prothrombinase activity supported by different sizes of PC/PS (80%/20% w/w) vesicles, as previously described. Large LMVs were prepared in TBS by vortexing a dried lipid suspension until all lipid was suspended.10 SUVs were prepared by sonication as described.1-3 LUVs were prepared by extruding LMV suspensions 20 times back and forth (total of 10 passes through the membrane) through a 0.1-μm polycarbonate membrane with a Mini-Extruder (Avanti Polar Lipids).10 (D) The effects of SkM, CM, or PC/PS LUV (PC/PS, 80%/20% w/w) on the initial rate of prothrombin (solid circles) or DG-prothrombin (open circles) activation by FXa and FVa were determined. Various concentrations of SkM, CM, or PC/PS (80%/20%) LUVs were incubated with FVa (2.4 nM final) and FXa (0.2 nM final) in TBSA plus 5 mM CaCl2. Thrombin generation was initiated by the addition of prothrombin or DG-prothrombin (0.75 μM final) and, after 10 minutes, was quenched by adding EDTA (10 mM final). The rate of thrombin formation was quantified by hydrolysis of thrombin fluorogenetic substrate. (E) FX activation assays were used to determine TF-like activity in SkM and CM preparations. Various concentrations of human or rabbit TF (Innovin, Dade or Pacific Hemostasis Thromboplastin-DS), SkM, or CM were mixed with FVIIa (0.5 nM final) in TBSA 5 mM CaCl2, and the addition of FX (0.25 μM final) initiated the reaction. After 10 minutes at room temperature, the addition of EDTA (10 mM final) quenched the reaction, and the rate of FXa formation was quantified by hydrolysis of an FXa chromogenic substrate (BIOPHEN CS11 [22]; Aniara, West Chester, OH). (F) The inhibitory effect of anti-FVII mAb on FX activation by FVIIa with human or rabbit TF was determined. Anti-FVII mAb 3G1215 (20 µg/mL final) was preincubated with FVIIa for 10 minutes at room temperature. Then TF (1.5 pM) with FX (final 250 nM) plus 5 mM CaCl2 was added and, after 10 minutes the reaction was quenched with EDTA (10 mM final). The rate of FXa formation was quantified by FXa activity (BIOPHEN CS11 [22]). (G) The effects of anti-FVII mAb or anti-TF mAb 3A5 on FX activation by FVIIa in the presence of rabbit TF, SkM, or CM was determined. Preincubation of anti-FVII mAb 3G12 (20 µg/mL, final) with FVIIa (0.5 nM final) for 10 minutes at room temperature was followed by the addition of FX (250 nM final) with rabbit TF (1.5 pM) or SkM (300 nM) or CM (50 nM) to start the reaction. After 10 minutes, the reaction was quenched by adding EDTA (10 mM final) and FXa activity (BIOPHEN CS11 [22]) was determined. For the anti-TF mAb experiment, preincubation of anti-TF mAb (RB TF 7-3A5)16 (20 µg/mL) with rabbit TF (1.5 pM) or SkM (300 nM) or CM (50 nM) for 10 minutes at room temperature was followed by the addition of FVIIa (0.5 nM final) and then FX (final 250 nM) in TBSA plus 5 mM CaCl2 to start the reaction. After 10 minutes, the reaction was quenched by adding EDTA (10 mM final) and FXa activity (BIOPHEN CS11 [22]) was determined. The 100% value was defined as FXa generated in the absence of added mAb. (H-J) The effects of anti-TF 3A5 mAb and anti-FVII 3G12 mAb on the procoagulant activity of SkM and CM in modified APTT plasma clotting assays that were previously described for the procoagulant activity of myosins.4 After preincubating SkM (H-I) or CM (J) or vehicle control buffer aliquots with anti-TF mAb (50 µg/mL) (H) or anti-FVII mAb (20 µg/mL) (I-J) for 10 minutes, the procoagulant activity was determined in the APTT assays. Each study included clotting assays for SkM and CM performed with (dashed lines) or without (solid lines) the addition of PC/PS (80%/20%) LUVs (1.0 µM final). For all studies in Figure 1 using SkM and CM, as for our published studies,1-5,17 the SkM preparation was dissolved and then dialyzed in Tris buffer (pH 7.4) containing 0.6 M NaCl at 4°C. After dialysis, particles causing turbidity were removed by high speed centrifugation (21 130g for 1 minute), which removed visible aggregates.
Effects of annexin V, anti-PS mAb, anti-TF mAb, and anti-FVII mAb on the procoagulant activities of SkM, CM, and PC/PS vesicles. (A-B) Annexin V (A) and anti-PS mAb 11.31 (B) were tested for inhibition of prothrombinase activity supported by SkM, CM, or PC/PS LUVs (PC/PS, 80%/20% w/w). Each inhibitor was incubated with FVa (2 nM, final) and FXa (0.24 nM, final) in Tris buffered saline (TBS) containing 0.5% bovine serum albumin (TBSA) and 5 mM CaCl2 in the presence of SkM (2.5 nM final), CM (2.5 nM final), or PC/PS vesicles (10 nM final). Addition of prothrombin (0.75 µM final) at room temperature initiated thrombin generation for 5 minutes that was quenched by EDTA (10 mM final). Thrombin activity was quantified by hydrolysis of the thrombin substrate (H-D-Phe-Pip-Arg-pNA). (C) Anti-PS mAb 11.31 was tested for inhibition of the prothrombinase activity supported by different sizes of PC/PS (80%/20% w/w) vesicles, as previously described. Large LMVs were prepared in TBS by vortexing a dried lipid suspension until all lipid was suspended.10 SUVs were prepared by sonication as described.1-3 LUVs were prepared by extruding LMV suspensions 20 times back and forth (total of 10 passes through the membrane) through a 0.1-μm polycarbonate membrane with a Mini-Extruder (Avanti Polar Lipids).10 (D) The effects of SkM, CM, or PC/PS LUV (PC/PS, 80%/20% w/w) on the initial rate of prothrombin (solid circles) or DG-prothrombin (open circles) activation by FXa and FVa were determined. Various concentrations of SkM, CM, or PC/PS (80%/20%) LUVs were incubated with FVa (2.4 nM final) and FXa (0.2 nM final) in TBSA plus 5 mM CaCl2. Thrombin generation was initiated by the addition of prothrombin or DG-prothrombin (0.75 μM final) and, after 10 minutes, was quenched by adding EDTA (10 mM final). The rate of thrombin formation was quantified by hydrolysis of thrombin fluorogenetic substrate. (E) FX activation assays were used to determine TF-like activity in SkM and CM preparations. Various concentrations of human or rabbit TF (Innovin, Dade or Pacific Hemostasis Thromboplastin-DS), SkM, or CM were mixed with FVIIa (0.5 nM final) in TBSA 5 mM CaCl2, and the addition of FX (0.25 μM final) initiated the reaction. After 10 minutes at room temperature, the addition of EDTA (10 mM final) quenched the reaction, and the rate of FXa formation was quantified by hydrolysis of an FXa chromogenic substrate (BIOPHEN CS11 [22]; Aniara, West Chester, OH). (F) The inhibitory effect of anti-FVII mAb on FX activation by FVIIa with human or rabbit TF was determined. Anti-FVII mAb 3G1215 (20 µg/mL final) was preincubated with FVIIa for 10 minutes at room temperature. Then TF (1.5 pM) with FX (final 250 nM) plus 5 mM CaCl2 was added and, after 10 minutes the reaction was quenched with EDTA (10 mM final). The rate of FXa formation was quantified by FXa activity (BIOPHEN CS11 [22]). (G) The effects of anti-FVII mAb or anti-TF mAb 3A5 on FX activation by FVIIa in the presence of rabbit TF, SkM, or CM was determined. Preincubation of anti-FVII mAb 3G12 (20 µg/mL, final) with FVIIa (0.5 nM final) for 10 minutes at room temperature was followed by the addition of FX (250 nM final) with rabbit TF (1.5 pM) or SkM (300 nM) or CM (50 nM) to start the reaction. After 10 minutes, the reaction was quenched by adding EDTA (10 mM final) and FXa activity (BIOPHEN CS11 [22]) was determined. For the anti-TF mAb experiment, preincubation of anti-TF mAb (RB TF 7-3A5)16 (20 µg/mL) with rabbit TF (1.5 pM) or SkM (300 nM) or CM (50 nM) for 10 minutes at room temperature was followed by the addition of FVIIa (0.5 nM final) and then FX (final 250 nM) in TBSA plus 5 mM CaCl2 to start the reaction. After 10 minutes, the reaction was quenched by adding EDTA (10 mM final) and FXa activity (BIOPHEN CS11 [22]) was determined. The 100% value was defined as FXa generated in the absence of added mAb. (H-J) The effects of anti-TF 3A5 mAb and anti-FVII 3G12 mAb on the procoagulant activity of SkM and CM in modified APTT plasma clotting assays that were previously described for the procoagulant activity of myosins.4 After preincubating SkM (H-I) or CM (J) or vehicle control buffer aliquots with anti-TF mAb (50 µg/mL) (H) or anti-FVII mAb (20 µg/mL) (I-J) for 10 minutes, the procoagulant activity was determined in the APTT assays. Each study included clotting assays for SkM and CM performed with (dashed lines) or without (solid lines) the addition of PC/PS (80%/20%) LUVs (1.0 µM final). For all studies in Figure 1 using SkM and CM, as for our published studies,1-5,17 the SkM preparation was dissolved and then dialyzed in Tris buffer (pH 7.4) containing 0.6 M NaCl at 4°C. After dialysis, particles causing turbidity were removed by high speed centrifugation (21 130g for 1 minute), which removed visible aggregates.
Annexin V potently inhibits the prothrombinase activity of SkM and CM, whereas an anti-PS mAb very weakly inhibits their prothrombinase activity. How might these data be interpreted? Annexin V binds not only PS but also to other anionic PLs (eg, phosphatidic acid [PA] and phosphatidylinositol [PI])11,12 in addition to anionic substances such as oleic acid and sodium dodecyl sulfate.13,14 Thus, anionic PLs and/or lipids could be involved in the procoagulant activity of SkM and CM. Anti-PS mAb 11.31 binds not only PS but also other anionic PLs such as phosphatidylglycerol, cardiolipin, PI, and PA.9 Assuming that the anti-PS mAb at very high concentrations inhibits SkM and CM prothrombinase activity because of its ability to recognize myosin-associated procoagulant lipid components with a much weaker affinity than for PS in vesicles, these annexin V and anti-PS mAb data (Figure 1A-C) support the interpretation that anionic PLs or anionic lipids, other than or in addition to PS, contribute to SkM and CM prothrombinase activity. This interpretation for the requirement for myosin-associated procoagulant anionic lipids is entirely consistent with the recent insightful report by Novakovic and Gilbert.6 This interpretation does not rule out minor contributions of PS to SkM and CM prothrombinase activity. But based on anti-PS mAb data, we propose that the enhancement of thrombin generation by SkM or CM is definitely not due to contaminating PS.6
Whether FXa directly binds only to contaminating PL vesicles6 or directly to the myosin protein can be inferred from reports1,3 showing that SkM and CM, but not PS-containing phospholipid vesicles, support the prothrombinase activity of DG-FXa, with each showing 30% activity vs <1% activity for PC/PS vesicles.1,3 In contrast to FXa, the Gla domain of prothrombin is required for any measurable prothrombinase activity supported by SkM, CM, or PC/PS vesicles (Figure 1D). Thus, because anionic PLs promote coagulation factor assemblies by binding Gla domains, we posit that myosin-associated anionic PLs bind the Gla domain of prothrombin.
Novakovic and Gilbert6 reported the presence of TF-like activity in SkM and CM preparations.6 When TF in myosin preparations was quantified using purified FX activation (tenase) assays, SkM had ∼3.4 pmol TF per µmol SkM and CM had ∼38 pmol TF per µmol (Figure 1E). To assess the relative importance of rabbit TF in SkM or bovine TF in CM for the procoagulant activities of each myosin in plasma clotting assays, we used mAbs that inhibit TF activity (Figure 1F-G). The anti-FVII mAb 3G12 blocked >90% of the tenase activity of human and rabbit TF (Figure 1F), as expected, whereas the anti-rabbit TF mAb (RbTF7 3A5) inhibited the tenase activity of rabbit TF and SkM, as expected, but not the tenase activity of bovine CM (Figure 1G). Modified activated partial thromboplastin time (APTT) clotting assays, which were previously used to characterize the procoagulant activity of SkM,4 were used here to assess the extent to which mAb blockade of TF-like activity ablates SkM and CM procoagulant activity in plasma. These APTT assays were performed with and without the addition of 1.0 µM PC/PS so that the relative influence of modest PS addition could be monitored. For SkM (Figure 1H), the anti-TF mAb had minimal effects, and for SkM and CM (Figure 1I-J), the anti-FVII mAb had minimal effects. So the presence or absence of low levels of anionic PLs or of TF had negligible effects on the procoagulant activity of SkM or CM in plasma clotting assays. Thus, SkM and CM exert significant procoagulant activity in plasma independent of contaminating TF or contaminating PS-containing PL vesicles, which disputes the previous hypothesis.6
In summary, the insightful reported ability of lipid-neutralizing reagents to inhibit the prothrombinase activities of SkM and CM6 plus our new data can be integrated with our published data1-4 to update our previous model for the prothrombinase activity of SkM.1 The updated model (Figure 2) for SkM and CM to enhance thrombin generation is centered on the existence of a myosin-lipid complex involving anionic PLs, most likely other than PS, which enhance prothrombin activation. This prothrombinase complex involves binding of FXa directly to myosin in its neck region as well as binding of FVa and prothrombin to the myosin-PL complex. For future studies, identification and characterization of the anionic PLs that help determine the enhancement of prothrombinase activity by SkM and CM may yield novel insights with translational implications.
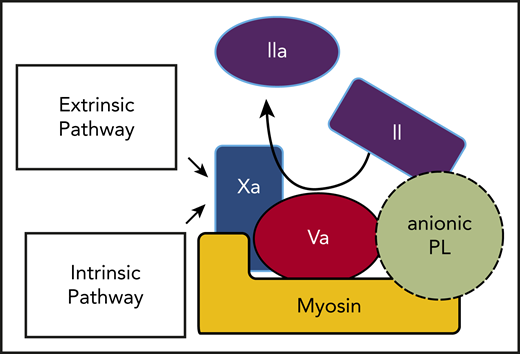
Thrombin generation can be driven by prothrombin activation on the surface of striated muscle myosin-anionic PL complexes that bind FXa and FVa. The intrinsic and extrinsic coagulation pathways converge at the generation of FXa, which is the key enzyme that activates prothrombin. As depicted, both FXa and its cofactor FVa bind to myosin-PL, which potently promotes prothrombin activation to generate thrombin. Prothrombin might bind to FVa and myosin and also to PLs via its Gla domain.
Thrombin generation can be driven by prothrombin activation on the surface of striated muscle myosin-anionic PL complexes that bind FXa and FVa. The intrinsic and extrinsic coagulation pathways converge at the generation of FXa, which is the key enzyme that activates prothrombin. As depicted, both FXa and its cofactor FVa bind to myosin-PL, which potently promotes prothrombin activation to generate thrombin. Prothrombin might bind to FVa and myosin and also to PLs via its Gla domain.
For original data, please contact John H. Griffin by e-mail at jgriffin@scripps.edu.
Acknowledgments
This work was supported by grants from the National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute (R01 HL133728 and R01 HL142975) (J.H.G.) and (UM1 HL120877) (W.R.), and the NIH/National Cancer Institute (U54 CA210181 and R01 CA243577) (R.A.B.).
Authorship
Contribution: S.M., J.A.F., and H.D. performed experiments; R.A.B. provided 11.31 mAb; W.R. provided anti-TF and anti-FVII mAbs; S.M., H.D., and J.H.G. designed the research and wrote the paper; and all authors analyzed results and read and approved the final manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: John H. Griffin, Department of Molecular Medicine IMM316, The Scripps Research Institute, 10550 N Torrey Pines Rd, La Jolla, CA 92037; e-mail: jgriffin@scripps.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal