

Key Points
FcRn participates in the induction of TF activity by IgG-containing ICs.
Inhibition of FcRn may reduce the prothrombotic phenotype of patients with disorders mediated by IgG-containing ICs.

Abstract
Thromboembolism complicates disorders caused by immunoglobulin G (IgG)–containing immune complexes (ICs), but the underlying mechanisms are incompletely understood. Prior evidence indicates that induction of tissue factor (TF) on monocytes, a pivotal step in the initiation, localization, and propagation of coagulation by ICs, is mediated through Fcγ receptor IIa (FcγRIIa); however, the involvement of other receptors has not been investigated in detail. The neonatal Fc receptor (FcRn) that mediates IgG and albumin recycling also participates in cellular responses to IgG-containing ICs. Here we asked whether FcRn is also involved in the induction of TF-dependent factor Xa (FXa) activity by IgG-containing ICs by THP-1 monocytic cells and human monocytes. Induction of FXa activity by ICs containing IgG antibodies to platelet factor 4 (PF4) involved in heparin-induced thrombocytopenia (HIT), β-2-glycoprotein-1 implicated in antiphospholipid syndrome, or red blood cells coated with anti-(α)-Rh(D) antibodies that mediate hemolysis in vivo was inhibited by a humanized monoclonal antibody (mAb) that blocks IgG binding to human FcRn. IgG-containing ICs that bind to FcγR and FcRn induced FXa activity, whereas IgG-containing ICs with an Fc engineered to be unable to engage FcRn did not. Infusion of an α-FcRn mAb prevented fibrin deposition after microvascular injury in a murine model of HIT in which human FcγRIIa was expressed as a transgene. These data implicate FcRn in TF-dependent FXa activity induced by soluble and cell-associated IgG-containing ICs. Antibodies to FcRn, now in clinical trials in warm autoimmune hemolytic anemia to lower IgG antibodies and IgG containing ICs may also reduce the risk of venous thromboembolism.
Introduction
Thromboembolism is a serious complication of disorders caused by immunoglobulin G (IgG)–containing immune complexes (ICs), including heparin-induced thrombocytopenia (HIT),1 antiphospholipid syndrome (APS),2,3 and warm autoimmune hemolytic anemia (WAHA).4-6 Multiple cellular pathways contribute to thromboembolism, including endothelial damage with loss of antithrombotic functions, generation of factor Xa (FXa) on monocytes, release of microparticles, elaboration of neutrophil extracellular traps, and activation of complement and diverse prothrombotic products released by damaged erythrocytes,7,8 among others.
Common to many prothrombotic pathways is the induction of tissue factor (TF), a pivotal step in the initiation, localization, and propagation of coagulation. Induction of TF activity by IgG-containing ICs is incompletely understood. In the case of HIT, antibodies to the IgG Fc receptor IIa (FcγRIIa; also known as CD32a) inhibit expression of TF on monocytes in vitro in response to PF4/polyanion IgG-containing ICs9,10 ; expression of FcγRIIa is required for development of thrombosis in a passive murine model of the disease,11 and the H/R131 polymorphism in FcγRIIa may influence the risk of thrombosis in vivo.10 Involvement of FcγRs has also been implicated in models of APS,12 in part by engaging toll-like receptor 2 (TLR-2) and TLR-4,13-15 among other pathways.2 However, it is unclear whether ICs promote TF expression exclusively through FcγRs or TLRs.
IgG-containing ICs can be modulated by the neonatal Fc receptor (FcRn),16,17 an ∼45-kDa nonpolymorphic major histocompatibility complex class I–like molecule that associates with β2-microglobulin.18 Binding of monovalent IgG to FcRn is pH dependent, occurring efficiently at acidic pH (pH <6.5) that develops in early sorting endosomes but not at neutral pH.19,20 FcRn binds at the CH2-CH3 interface of the IgG Fc domain, which is distinct from the binding site for classical FcγRs.21-23 This pH-dependent reversible binding of IgG underlies an important function of FcRn, involving the transport of maternal IgG across the placenta to the fetus and recycling of plasma IgG by endothelial and hematopoietic cells.24,25 FcRn also protects IgG-containing ICs from catabolism via mechanisms that are poorly understood but likely involve intracellular retention of internalized IgG-containing ICs16,19,26 involved in the pathogenesis of autoimmune diseases.27-30
IgG-containing ICs affect the behavior of diverse hematopoietic cells, including neutrophils, monocytes, macrophages, dendritic cells, and B cells, that express both classical FcγRs (FcγRI, FcγRIIa, FcγRIIb, and FcγRIII) and FcRn.17,19,31 Expression of FcRn on hematopoietic cells not only controls the half-life of IgG-containing ICs in mice19 but also modulates their ability to produce innate cytokines and engage in antigen presentation and cross-presentation to CD4+ and CD8+ T cells, respectively.16,17 Recently, these observations have been extended to humans who have been given a therapeutic monoclonal antibody (mAb) MoAb, SYNT001, that disrupts binding of IgG to FcRn, blocks recycling of IgG antibodies, promotes clearance of IgG-containing ICs from the circulation, and blocks the ability of IgG-containing ICs to induce innate and adaptive immune responses.17 In light of these findings, we investigated whether FcRn also regulates the induction of TF on a monocyte-like cell line and on human monocytes by soluble and particulate ICs involved in HIT, APS, and WAHA. These results have important clinical implications by suggesting inhibition of FcRn function may be used to attenuate the risk of thromboembolic complications in affected patients.
Materials and methods
Cells and reagents
Antibodies
An IgG-blocking antibody to TF was obtained from American Diagnostica (Stamford, CT), an IgG control and α-human F(ab)′2 antibody from Jackson ImmunoResearch Laboratories (West Grove, PA), and an IgG α-tissue factor pathway inhibitor (TFPI)-β antibody from Abcam (Cambridge, MA). The human IgG mAb α-Rh(D) BRAD-3 and human IgM α-Rh(D) mAb MAD-2 were from the International Blood Group Reference Laboratory (Bristol, United Kingdom). The human IgG polyclonal α-Rh(D) antibody RhoGAM was from Kendrion Biopharma (Fort Lee, NJ). KKO is a murine HIT-like α-PF4/heparin mAb.11,34 A chimeric α-NIP human IgG1 mAb that is bound by both FcγR and FcRn (designated IgGWT) and a mutated α-NIP human IgG1 mAb (I253A, H310A, H435A; designated IgGIHH) that retains binding to FcγR but not to FcRn have been characterized previously.19 Human IgG α-β2GP1 antibodies were isolated from the plasma of 2 patients with APS, and rabbit IgG α-β2GPI was generated, isolated, and characterized as described.33,35 Human IgG was isolated from the plasma of 3 patients with HIT.9 A function-blocking rabbit polyclonal antibody recognizing the first 2 Kunitz domains of TFPI was produced by Genemed Synthesis (San Antonio, TX).36 F(ab)′2 fragments of the α-mouse CD41 mAb (BD Biosciences; MWReg30) were used to detect murine platelets in the cremaster laser injury model. A blocking mAb to human FcγRIIa (clone IV.3) was purified as described.36 The phycoerythrin (PE)-conjugated FUN-2 mAb (product 303206; BioLegend, San Diego, CA) is an IgG2b antibody that recognizes FcγIIa (CD32). The α-fibrin mAb (clone 59D8) was generously provided by Hartmut Weiler of the Blood Center of Wisconsin (Milwaukee, WI).37 SYNT001 is humanized affinity-matured deimmunized IgG4κ mAb containing an S241P mutation17 provided by Syntimmune (subsidiary of Alexion Pharmaceuticals, Inc.). DVN24 is an IgG2a murine α-mouse/human FcRn mAb.38 An irrelevant human IgG4 (S241P) and mouse IgG2a were used as the isotype controls for SYNT001 and DVN24, respectively. An α-human CD14 mAb conjugated with PE-Cy7 (product 301814) was from BioLegend. ADM31, an IgG2b mAb that recognizes the albumin binding site on FcRn distinct from the site recognized SYNT00139 was custom conjugated with PE by BioLegend. A PE-conjugated irrelevant mouse IgG2b was used as the isotype control (product 402204; BioLegend). PE-conjugated anti-human CD142 (product 550312; BD Pharmingen) was used to detect expression of TF protein on human monocytes.
ICs
Five sets of ICs were generated: (1) PF4 (0-10 µg/mL) and KKO or (2) human HIT IgGs (50 µg/mL), (3) β2GPI (0-10 µg/mL) and human or rabbit IgG α-β2GPI (50 µg/mL), (4) Rh(D)+ or Rh(D)− red blood cells (RBCs) and either murine monoclonal or human polyclonal IgG and IgM α-Rh(D) antibodies, and (5) NIP-conjugated ovalbumin (OVANIP) containing ∼15 NIP molecules (200 µg/mL) was incubated with either IgGWT or IgGIHH monomeric mAb (10 µg/mL) for 1 hour at 37°C to form ICs, and one-tenth of the reaction volume was add to the cell suspension.
Induction of TF-dependent FXa activity by soluble ICs
THP-1 cells were cultured in RPMI 1640 supplemented with 10% fetal bovine serum (FBS), 4.5 mg/mL of glucose, 1 mM of sodium pyruvate, 2 mM of l-glutamine, 100 U/mL of penicillin, 100 µg/mL of streptomycin, and 0.25 µg/mL of amphotericin B. Cells kept at a density of 0.25 × 106/mL to 1.0 × 106/mL at 37°C under 5% CO2 were plated within wells of a V-shaped 96-well plate in a volume of 100 µL in RPMI 1640/5% FBS (final, 105 cells per well) or in serum-free media where indicated. Cell-associated ICs were formed between PF4 and KKO and between β2GPI and human or rabbit IgG α-β2GP1 antibodies by adding each component separately to the cell suspension. In other experiments, preformed soluble complexes between OVANIP and IgGWT or IgGIHH were added to cell suspensions. Cells incubated with each antigen or antibody alone served as controls. Cells were incubated with ICs or controls for 0 to 3 hours, washed twice by centrifugation at 277 g in RPMI/5% FBS, resuspended in 100 µL of RPMI/5% FBS, and incubated overnight. Then, 100 µL of complete RPMI 1640 (10% FBS) was added to each well, cells were thoroughly resuspended, and aliquots from the cell suspensions were analyzed for FXa activity. Cells were incubated for an additional 18 hours, and 100 µL of complete RPMI 1640/10% FBS was added to each well, and the cells were thoroughly resuspended. The FcRn-blocking mAb SYNT001 or control IgG4 (0-200 µg/mL each) was added to the THP-1 cells for 30 minutes before ICs. In some experiments, human umbilical vein endothelial cells (HUVECs; passage 3; 7-10 000 cells per well) were incubated with IgGWT IC and IgGIHH IC for 3 or 18 hours or with SYNT001 (200 μg/mL) for 1 hour. Cells were washed 3 times with 50% growth medium; α-human F(ab)′2 (50 µg/mL) was added for 15 hours to cross-link FcRn, the cells were washed, and FXa activity was measured. The methods used to measure FXa activity by human monocytes are described in detail in the data supplement.
Induction of FXa by particulate ICs
Panoscreen I Rh(D)+ or Panoscreen III Rh(D)− reagent RBCs (2 × 107) were incubated with mAbs BRAD-3 or MAD-2 (100 ng/mL) or with the polyclonal antibody RhoGAM at 37°C for 30 minutes, washed, and resuspended in phosphate-buffered saline (PBS). Antibody-sensitized or control (PBS alone) RBCs (1 × 106) were added to 105 THP-1 cells per well for 3 hours at 37°C under 5% CO2. Cells were washed twice, resuspended in 100 µL of media containing 5% FBS, and incubated under the same conditions overnight, and FXa activity was measured. SYNT001 or control IgG4 (0-200 µg/mL each) was added to the THP-1 cells for 30 minutes before adding IgG antibody-coated RBCs.
Measurement of FXa activity, TF protein, TF messenger RNA, TFPI, and subcellular localization
These methods are described in detail in the supplemental Materials (available on the Blood Web site).
Interaction of SYNT001 with FcRγIIa
The methods used to study the potential for SYNT001 to interact with FcRγIIA are described in detail in the supplemental Materials.
Membrane colocalization
To examine colocalization of FcRn with PF4/KKO ICs and/or FcγRIIa, THP-1 cells were plated in 8-well chamber slides (LabTek, Campbell, CA), precoated for 1 hour at 37°C with RetroNectin (100 μg/mL; TaKaRa Bio, Mountain View, CA), and allowed to adhere for 16 to 18 hours in growth media. Adherent THP-1 cells were incubated with PF4/KKO ICs or with buffer alone for 15 minutes, washed in PBS, fixed with 4% paraformaldehyde in PBS for 15 minutes at room temperature, and washed with PBS; unreactive sites were blocked with 1% bovine serum albumin. Human α-FcRn antibody (SYNT001), mouse α-human PF4/heparin mAb (KKO) or mouse control α-human PF4 monomer mAb (RTO),40 and mouse α-human FcγRIIa MoAb (clone IV.3) and the corresponding control IgGs (ie, human IgG4 as the negative control for SYNT001 and mouse IgG [TRA] for KKO and for IV.3) were conjugated with Alexa 488, Alexa 568, and Alexa 647 fluorescent dyes, respectively, using antibody-labeling kits (Thermo Fisher, Invitrogen).41 Cells exposed to HIT ICs or controls were incubated with Alexa 488/SYNT001, Alexa 568/KKO, and Alexa 647/IV.3 mAbs for 1 hour at room temperature. Alexa 488–conjugated human IgG4 and Alexa 568– or Alexa 647–conjugated isotype-matched mouse-irrelevant IgG2b served as the negative controls. In other experiments, THP-1 cells were preincubated with SYNT001 or control human IgG4 (200 µg/mL), with IV.3 (200 µg/mL), or with control mouse IgG for 30 minutes before adding PF4/KKO ICs for 15 minutes. The cells were washed, fixed, and stained as above. Cells were mounted in ProLong Gold Antifade Mountant, stained with DAPI (Thermo Fisher Scientific/Molecular Probes, Eugene, OR), and examined using a confocal laser-scanning microscope (Zeiss LSM 710; Carl Zeiss, Heidelberg, Germany) equipped with a Plan Apo 40× water-immersion objective lens (NA 1.2). The Z-stack distance between the slices was set as 0.3 µm, with a 1024 × 1024 pixel resolution for each slice. Three-dimensional reconstruction and maximal projection were performed using Volocity 6.3 software (Perkin Elmer, Waltham, MA).
In vivo studies
Thrombus formation
Mice expressing platelet-specific human PF4 (hPF4+) and the human FcγRIIa-R131 isoform (FcγRIIa+)42 on a Cxcl4−/− background43 were studied. Six- to 10-week-old male mice were injected intraperitoneally with α-mouse FcRn (DVN24) or control IgG2a (200 µg per animal) 24 hours and 1 hour before laser injury. Arterioles (20-40 μm in diameter) were selected for examination. Vascular injury was induced with an SRS NL100 pulsed nitrogen dye laser (440 nm) focused on the vessel wall through the microscope objective. The laser was pulsed until the vessels were perforated and a small number of RBCs escaped. α-mouse CD41, α-fibrin, and KKO were infused as 100-μL boluses via a catheter placed into the jugular vein 10 minutes before the initial injury. Intravital microscopy and data collection were performed as described.44 Widefield time-lapsed images of platelet and fibrin accumulation were analyzed using Slidebook 6.0 (Intelligent Imaging Innovations). Four to 10 injuries per mouse over a maximum experimental time of 1 hour were studied.
FXa activity
The method used to study the effect of FcRn blockade on FXa expression by monocytes from HIT mice is described in the supplemental Materials.
Statistical analysis
All data are presented as the mean ± standard error of the mean of at least 3 separate experiments. Differences between groups were calculated using GraphPad Prism 7 and tested for statistical significance using Student t test with Welch’s correction for unequal variances when necessary, or 1-way analysis of variance with indicated multiple comparison testing. Statistical significance was set at P < .05.
Results
Induction of FXa activity by soluble IgG-containing ICs
PF4 binds to glycosaminoglycans on the plasma membranes of human monocytes, forming cell-surface ICs recognized by the pathogenic murine HIT-like mAb KKO.34 Binding of KKO to human monocytes induces TF-dependent FXa activity, which has been shown to be dependent on FcγRIIa.11 Consistent with these prior observations, addition of KKO plus PF4 (HIT ICs) induced the expression of FXa activity on monocytic THP-1 cells measured by factor VIIa– and FX-dependent cleavage of an FXa chromogenic substrate (Figure 1A). Formation of the HIT antigen between PF4 and glycosaminoglycans followed a parabolic concentration-dependent pattern.45 In line with this, generation of FXa also depended on the ratio of PF4 to KKO and the duration of exposure to the IgG-containing ICs (data not shown). Under optimal conditions for HIT IC formation, exposure of THP-1 cells to KKO plus PF4 increased the initial velocity of FXa generation 2.9- ± 0.5-fold (ie, from 35 ± 4 pM per minute per 5000 cells to 104 ± 20 pM per minute per 5000 cells; P = .001; n = 5), whereas PF4 alone did not induce increased FXa expression significantly (42 ± 5 pM per minute per 5000 cells). Identical results were seen when THP-1 cells were stimulated in serum-containing and serum-free media, excluding a contribution of exogenous TF-independent coagulation factors (data not shown). HIT ICs also stimulated the generation of FXa activity by isolated human monocytes 7.7- ± 0.7-fold (P < .04; n = 3; supplemental Figure 1B). Incubation with HIT ICs for 1, 3, and 24 hours increased TF messenger RNA vs the effect of PF4 1.9- ± 0.06-fold (P = not significant), 3.3- ± 0.5-fold (P = .012), and 7.9- ± 0.2-fold (P = 2.8e−6; n = 6 for each condition), respectively, compared with cells incubated with PF4 alone at the same time points (supplemental Figure 1C), and cell-surface TF protein assessed by flow cytometry was increased 3.2- ± 0.6-fold (P = .02; n = 8).
![Induction of TF activity in response to IgG immune complexes. (A) Induction of TF by HIT ICs. THP-1 cells were incubated with buffer, PF4 (10 µg/mL), or PF4 plus KKO (50 µg/mL) for 3 hours in these and all experiments that follow unless indicated otherwise. The cells were washed free of unbound ICs and incubated overnight, and TF expression by the cell suspension was measured by the generation of FXa activity. Data from 5 independent experiments (mean ± standard error of the mean [SEM]) are shown as fold increase in the maximal velocity of FXa generation relative to cells incubated with PF4 alone, designated 1. (B). Subcellular distribution of TF activity induced by HIT ICs. THP-1 cells were incubated with PF4 and KKO, and total cell-associated TF activity in the cell suspension was determined as in panel A (cells). Aliquots from these same cell suspensions were centrifuged at 1200 g for 10 minutes, and the supernatants were collected (Sup 1). Aliquots from Sup 1 were centrifuged at 21 000 g for 20 minutes and again collected (Sup 2). TF activity in the 3 fractions was measured concurrently. Data shown are mean ± SEM of 4 independent experiments relative to cells stimulated by PF4 (10 μg/mL) alone. (C) Effect of anti-TF antibody (α-TF Ab) on FXa activity induced by HIT ICs. THP-1 cells were stimulated with PF4 and KKO in the presence of IgG α-TF Ab or control IgG (Con Ab 20 µg/mL each), and cell-associated TF activity was measured as in panel A. Data from 5 independent experiments (mean ± SEM) are shown relative to cells stimulated by PF4 (10 μg/mL) alone. (D) Effect of α-TFPI-α antibody on FXa activity induced by HIT ICs. THP-1 cells were stimulated with PF4 and KKO in the presence (+) or absence (−) of α-TFPI Ab (50 µg/mL) for 24 hours, and FXa activity was measured as in panel A. Data from 5 independent experiments (mean ± SEM) are shown relative to cells stimulated by PF4 (10 μg/mL) alone. †P = .001 by Student t test with Welch’s correction; $P = .005 by 1-way analysis of variance (ANOVA) with Sidak’s multiple comparison test; &P = .0178, #P = .0001, and ***P < .0001 by 1-way ANOVA with Sidak’s multiple comparison test; @P = .0468 by Student t test with Welch’s correction.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/23/10.1182_blood.2019001133/2/m_bloodbld2019001133f1.png?Expires=1767721401&Signature=sT-4M5xrf-yhCv4~eIX9GAJl9VhuHv7zoYreafTaVNdS54XwQXGEy0eik1s~I1cbvB0oKCT-uUZxCQJUdozZ6cOgUUqoZF~sVVdiuFGpxh2PThC2WoJwTJxZL-eAqS7-76wcyQC-w004Gw~VnxKHfFzjilLTcNuI7J4GLviAYnJseTndmBhaVx3InZRqOxDTgHdYn9Q9KJTPU1boG6lWSEI7acs9YM9VbKbSJ-Ebx8TPXb9CaT9k39KR2NwLYIseDHBMC-MsFzKRMMoDohJdG29yKpZF0XuDtY2-lnX41AHpRGOoFsRcMK4GfQT2SgEbHWmaqm63vC8kepRtlr4qPw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Induction of TF activity in response to IgG immune complexes. (A) Induction of TF by HIT ICs. THP-1 cells were incubated with buffer, PF4 (10 µg/mL), or PF4 plus KKO (50 µg/mL) for 3 hours in these and all experiments that follow unless indicated otherwise. The cells were washed free of unbound ICs and incubated overnight, and TF expression by the cell suspension was measured by the generation of FXa activity. Data from 5 independent experiments (mean ± standard error of the mean [SEM]) are shown as fold increase in the maximal velocity of FXa generation relative to cells incubated with PF4 alone, designated 1. (B). Subcellular distribution of TF activity induced by HIT ICs. THP-1 cells were incubated with PF4 and KKO, and total cell-associated TF activity in the cell suspension was determined as in panel A (cells). Aliquots from these same cell suspensions were centrifuged at 1200 g for 10 minutes, and the supernatants were collected (Sup 1). Aliquots from Sup 1 were centrifuged at 21 000 g for 20 minutes and again collected (Sup 2). TF activity in the 3 fractions was measured concurrently. Data shown are mean ± SEM of 4 independent experiments relative to cells stimulated by PF4 (10 μg/mL) alone. (C) Effect of anti-TF antibody (α-TF Ab) on FXa activity induced by HIT ICs. THP-1 cells were stimulated with PF4 and KKO in the presence of IgG α-TF Ab or control IgG (Con Ab 20 µg/mL each), and cell-associated TF activity was measured as in panel A. Data from 5 independent experiments (mean ± SEM) are shown relative to cells stimulated by PF4 (10 μg/mL) alone. (D) Effect of α-TFPI-α antibody on FXa activity induced by HIT ICs. THP-1 cells were stimulated with PF4 and KKO in the presence (+) or absence (−) of α-TFPI Ab (50 µg/mL) for 24 hours, and FXa activity was measured as in panel A. Data from 5 independent experiments (mean ± SEM) are shown relative to cells stimulated by PF4 (10 μg/mL) alone. †P = .001 by Student t test with Welch’s correction; $P = .005 by 1-way analysis of variance (ANOVA) with Sidak’s multiple comparison test; &P = .0178, #P = .0001, and ***P < .0001 by 1-way ANOVA with Sidak’s multiple comparison test; @P = .0468 by Student t test with Welch’s correction.
Induction of TF activity in response to IgG immune complexes. (A) Induction of TF by HIT ICs. THP-1 cells were incubated with buffer, PF4 (10 µg/mL), or PF4 plus KKO (50 µg/mL) for 3 hours in these and all experiments that follow unless indicated otherwise. The cells were washed free of unbound ICs and incubated overnight, and TF expression by the cell suspension was measured by the generation of FXa activity. Data from 5 independent experiments (mean ± standard error of the mean [SEM]) are shown as fold increase in the maximal velocity of FXa generation relative to cells incubated with PF4 alone, designated 1. (B). Subcellular distribution of TF activity induced by HIT ICs. THP-1 cells were incubated with PF4 and KKO, and total cell-associated TF activity in the cell suspension was determined as in panel A (cells). Aliquots from these same cell suspensions were centrifuged at 1200 g for 10 minutes, and the supernatants were collected (Sup 1). Aliquots from Sup 1 were centrifuged at 21 000 g for 20 minutes and again collected (Sup 2). TF activity in the 3 fractions was measured concurrently. Data shown are mean ± SEM of 4 independent experiments relative to cells stimulated by PF4 (10 μg/mL) alone. (C) Effect of anti-TF antibody (α-TF Ab) on FXa activity induced by HIT ICs. THP-1 cells were stimulated with PF4 and KKO in the presence of IgG α-TF Ab or control IgG (Con Ab 20 µg/mL each), and cell-associated TF activity was measured as in panel A. Data from 5 independent experiments (mean ± SEM) are shown relative to cells stimulated by PF4 (10 μg/mL) alone. (D) Effect of α-TFPI-α antibody on FXa activity induced by HIT ICs. THP-1 cells were stimulated with PF4 and KKO in the presence (+) or absence (−) of α-TFPI Ab (50 µg/mL) for 24 hours, and FXa activity was measured as in panel A. Data from 5 independent experiments (mean ± SEM) are shown relative to cells stimulated by PF4 (10 μg/mL) alone. †P = .001 by Student t test with Welch’s correction; $P = .005 by 1-way analysis of variance (ANOVA) with Sidak’s multiple comparison test; &P = .0178, #P = .0001, and ***P < .0001 by 1-way ANOVA with Sidak’s multiple comparison test; @P = .0468 by Student t test with Welch’s correction.
Most FXa activity (67.8% ± 1.1%; P < .03) was associated with the cell surface, with the remainder in the fluid phase after a 1200-g centrifugation, consistent with released extracellular vesicles (Figure 1B). Little FXa activity remained in the fluid phase after a 21000-g centrifugation of extracellular vesicle–containing supernatants, excluding a major contribution by exosomes and other submicron-sized particles (Figure 1B).
Generation of FXa activity induced by HIT ICs was completely abolished by an α-TF antibody (Figure 1C). The α-TF antibody also reduced TF expression by control THP-1 cells as well, as expected. Although greater FXa activity was measured in the presence of a polyclonal antibody that inhibited TFPI activity (Figure 1D), neither total TFPI nor TFPI-α was detected in cell lysates by enzyme-linked immunosorbent assay, nor was TFPI-β detected in supernatants after addition of phosphatidylinositol-phospholipase C. Thus, TFPI remained below its detection limit of 60 pg/mL. This indicates that the increase in FXa generation was attributable to induction of TF activity rather than decreased inhibition of FXa activity by TFPI.
Involvement of FcRn in in vitro induction of TF-dependent FXa activity
To examine the role of FcRn in the induction of FXa activity by IgG-containing ICs, we first examined the effect of blocking the receptor. To do so, we added α-human FcRn mAb SYNT001 that blocks FcRn-IgG interactions or an irrelevant human IgG4 isotype control to THP-1 cells for 30 minutes before adding HIT ICs and measured FXa activity. SYNT001 inhibited the induction of FXa activity by HIT ICs in a dose-dependent manner. FXa activity was inhibited by 59.9% ± 10.4% (P = .005) and 62.8% ± 14.7% (P = .0006) by SYNT001 at concentrations of 100 and 200 µg/mL, respectively, as compared with 200 μg/mL of control IgG4 based on calculations of initial rate constants (data not shown) or as fold increase in activity (Figure 2A). SYNT001 (200 µg/mL) also significantly inhibited the induction of FXa activity on THP-1 cells by each of 3 human HIT IgG antibodies (supplemental Figure 1A) and inhibited FXa generation induced by PF4/KKO on human monocytes by 70.9% ± 7.6% (P = .039; supplemental Figure 1B). In contrast, SYNT001 had no effect on FXa activity induced by 10 µg/mL of LPS (data not shown). Moreover, SYNT001 itself did not induce FXa activity on THP-1 cells or HUVECs, even at the highest concentrations tested (200 µg/mL), or when it was cross-linked by adding α-human F(ab)′2 antibody (data not shown). The inhibition of FXa activity was not simply the result of SYNT001 binding to FcRn via its F(ab) domain or to FcγRIIa via its Fc domain, because binding was not increased when expression of FcγRIIa was increased >30-fold (supplemental Figure 2).
![Inhibition of TF expression by α-FcRn antibody. (A) HIT ICs. THP-1 cells were stimulated by HIT ICs as in Figure 1 in the presence of an IgG4 α-FcRn mAb (200, 100, or 50 µg/mL) or an IgG4 control immunoglobulin (Con Ab; 200 µg/mL), and TF activity was measured as in Figure 1A. Data from 3 to 6 independent experiments (mean ± standard error of the mean [SEM]) are shown relative to cells stimulated by PF4 (10 µg/mL) alone. (B) TF activity by cells stimulated with FcRn binding and nonbinding IgG ICs. To form ICs, OVANIP (200 µg/mL) was incubated with an α-NIP IgG antibody (10 µg/mL) that binds FcRn (IgGWT IC) or an isotype-matched antibody with mutations that permit binding to classical IgG Fc receptors but not FcRn (IgGIHH IC). The complexes were diluted 1:10 before addition to cells. Induction of TF activity in the absence or presence of α-FcRn antibody was determined as in panel A. Data from 3 independent experiments (mean ± SEM) are shown relative to cells stimulated with IgGIHH IC and α-FcRn. %P = .0124, ∧P = .0021, and ‡P = .0006 by 1-way analysis of variance (ANOVA) with Fisher’s least significant difference test; **P < .001 by 1-way ANOVA with Sidak’s multiple comparison test.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/23/10.1182_blood.2019001133/2/m_bloodbld2019001133f2.png?Expires=1767721401&Signature=0dFRe1X9S3IqoDbdI2gwD3-o0fB-WOJl2kCfK7qactbXfmdC6NdrU-a2evHmgpHjeMuN5YAIadWidpswjVZWUGflAaHj3LslMHhpQ4KL2En1EMoVIdsf4cfeTT0ub8VH9MjfO5WUEkI7re4g8LZ7~hZLm4li-qn9wbEelsPweOpSL4D3qYVg0JnJIWPKIJEQzzvJkg0uDZbcFiSbW3UDWiivS5mTPjlLHpcwaz9iqEDc9RxoRl60ZFtOedIPlp9yXSibOaD6-hQ0B~ZsuAUcp3HsgXB0jnypS0teCwdVP~mXsLp8zi6qroSTH4Paana9S0DboLuZAj32xN~bhigMog__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Inhibition of TF expression by α-FcRn antibody. (A) HIT ICs. THP-1 cells were stimulated by HIT ICs as in Figure 1 in the presence of an IgG4 α-FcRn mAb (200, 100, or 50 µg/mL) or an IgG4 control immunoglobulin (Con Ab; 200 µg/mL), and TF activity was measured as in Figure 1A. Data from 3 to 6 independent experiments (mean ± standard error of the mean [SEM]) are shown relative to cells stimulated by PF4 (10 µg/mL) alone. (B) TF activity by cells stimulated with FcRn binding and nonbinding IgG ICs. To form ICs, OVANIP (200 µg/mL) was incubated with an α-NIP IgG antibody (10 µg/mL) that binds FcRn (IgGWT IC) or an isotype-matched antibody with mutations that permit binding to classical IgG Fc receptors but not FcRn (IgGIHH IC). The complexes were diluted 1:10 before addition to cells. Induction of TF activity in the absence or presence of α-FcRn antibody was determined as in panel A. Data from 3 independent experiments (mean ± SEM) are shown relative to cells stimulated with IgGIHH IC and α-FcRn. %P = .0124, ∧P = .0021, and ‡P = .0006 by 1-way analysis of variance (ANOVA) with Fisher’s least significant difference test; **P < .001 by 1-way ANOVA with Sidak’s multiple comparison test.
Inhibition of TF expression by α-FcRn antibody. (A) HIT ICs. THP-1 cells were stimulated by HIT ICs as in Figure 1 in the presence of an IgG4 α-FcRn mAb (200, 100, or 50 µg/mL) or an IgG4 control immunoglobulin (Con Ab; 200 µg/mL), and TF activity was measured as in Figure 1A. Data from 3 to 6 independent experiments (mean ± standard error of the mean [SEM]) are shown relative to cells stimulated by PF4 (10 µg/mL) alone. (B) TF activity by cells stimulated with FcRn binding and nonbinding IgG ICs. To form ICs, OVANIP (200 µg/mL) was incubated with an α-NIP IgG antibody (10 µg/mL) that binds FcRn (IgGWT IC) or an isotype-matched antibody with mutations that permit binding to classical IgG Fc receptors but not FcRn (IgGIHH IC). The complexes were diluted 1:10 before addition to cells. Induction of TF activity in the absence or presence of α-FcRn antibody was determined as in panel A. Data from 3 independent experiments (mean ± SEM) are shown relative to cells stimulated with IgGIHH IC and α-FcRn. %P = .0124, ∧P = .0021, and ‡P = .0006 by 1-way analysis of variance (ANOVA) with Fisher’s least significant difference test; **P < .001 by 1-way ANOVA with Sidak’s multiple comparison test.
Second, THP-1 cells were incubated with model ICs composed of OVANIP and α-NIP IgGWT antibodies able to interact with both FcγR and FcRn or IgGIHH antibodies unable to interact with FcRn selectively.16 IgGWT ICs induced FXa activity on isolated THP-1 cells, whereas IgGIHH ICs did not (supplemental Figure 2B). IgGWT ICs also amplified the expression of TF by human monocytes compared with IgGIHH ICs (supplemental Figure 3). In contrast, neither IgGWT nor IgGIHH ICs induced FXa activity by HUVECs (data not shown). Because HUVECs express FcRn, but not other FcγRs, this suggests that engagement of FcRn alone on endothelial cells is not sufficient to generate FXa activity, in contrast to monocytes, which express both types of receptors.
Third, we asked whether HIT ICs colocalize with FcRn on THP-1 cells. In the absence of HIT ICs, FcRn was expressed in a diffuse pattern on the surface of THP-1 cells (Figure 3A upper Alexa-488–α-FcRn panel), consistent with previous reports that FcRn is expressed on human monocytes.31 FcγRIIa, as defined by the IV.3 antibody, was observed in small, diffuse clusters (Figure 3A upper Alexa-647–α-FcγRIIa panel).46,47 When THP-1 cells were incubated with HIT ICs for 15 minutes, FcRn and FcγRIIa redistributed into discrete clusters in proximity to KKO/PF4 (Figure 3A lower overlay + DAPI panel; supplemental Figure 1). SYNT001 totally prevented redistribution of FcRn into clusters containing HIT ICs and FcγRIIa and partially inhibited appearance of HIT ICs within clusters containing FcγRIIa (Figure 3B second row). Control IgG4 neither inhibited binding of HIT ICs nor altered the distribution of FcγRIIa or FcRn (Figure 3B upper panels). Preincubation with the α-FcγRIIa–blocking mAb IV.3 also partially inhibited binding of HIT ICs as previously reported32 and blocked the appearance of FcγRIIa in clusters containing HIT ICs and FcRn (Figure 3B third row). Preincubation of THP-1 cells with α-FcRn (SYNT001) and α-FcγRIIa antibodies (IV.3) completely blocked binding of HIT ICs (Figure 3B fourth row). Together, these results suggest that HIT ICs induce coclustering of FcRn with FcγRIIa under conditions that generate TF activity.

Colocalization of HIT ICs, FcRn, and FcγRIIa on THP-1 cells upon treatment with IgG-containing ICs. (A) THP-1 cells immobilized on RetroNectin were left untreated (top) or incubated with PF4 + KKO complexes (bottom) for 15 minutes as in Figure 1A, washed, and fixed in 4% paraformaldehyde in PBS. (B) Inhibition of HIT ICs binding and coclustering with FcRn and FcγRIIa by α-FcRn and α-FcγRIIa mAbs on the surface of THP-1 cells. THP-1 cells were preincubated with control human IgG4 and control mouse IgG (TRA; 200 μg/mL both; top), α-FcRn antibody (Ab; 200 μg/mL of SYNT001; second row) or with α-FcγRIIa Ab (200 μg/mL IV.3; third row) or with both Abs (bottom) for 30 minutes before addition of PF4 and KKO ICs for 15 minutes. The cells were then washed, fixed, and stained. PF4/KKO ICs, FcRn, and FcγRIIa were detected using Alexa-568–conjugated α-PF4/heparin mAb KKO (red), Alexa-488–conjugated α-human FcRn mAbs (green), and Alexa-647–conjugated FcγRIIa mAbs (pseudocolored in white). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; blue). Individual red, green, and deep-red channels and their overlays with blue channel (nuclei staining; overlay + DAPI) are shown. Staining with negative control Abs is shown within the insets in each panel. Scale bars, 10 μm.
Colocalization of HIT ICs, FcRn, and FcγRIIa on THP-1 cells upon treatment with IgG-containing ICs. (A) THP-1 cells immobilized on RetroNectin were left untreated (top) or incubated with PF4 + KKO complexes (bottom) for 15 minutes as in Figure 1A, washed, and fixed in 4% paraformaldehyde in PBS. (B) Inhibition of HIT ICs binding and coclustering with FcRn and FcγRIIa by α-FcRn and α-FcγRIIa mAbs on the surface of THP-1 cells. THP-1 cells were preincubated with control human IgG4 and control mouse IgG (TRA; 200 μg/mL both; top), α-FcRn antibody (Ab; 200 μg/mL of SYNT001; second row) or with α-FcγRIIa Ab (200 μg/mL IV.3; third row) or with both Abs (bottom) for 30 minutes before addition of PF4 and KKO ICs for 15 minutes. The cells were then washed, fixed, and stained. PF4/KKO ICs, FcRn, and FcγRIIa were detected using Alexa-568–conjugated α-PF4/heparin mAb KKO (red), Alexa-488–conjugated α-human FcRn mAbs (green), and Alexa-647–conjugated FcγRIIa mAbs (pseudocolored in white). Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; blue). Individual red, green, and deep-red channels and their overlays with blue channel (nuclei staining; overlay + DAPI) are shown. Staining with negative control Abs is shown within the insets in each panel. Scale bars, 10 μm.
Involvement of FcRn in induction of TF activity in response to other soluble or particulate ICs
We then asked if the involvement of FcRn was restricted to the ultralarge ICs formed by PF4 with HIT antibodies.32 To address this question, we first incubated THP-1 cells with ICs formed by adding human β2GP1 and an IgG rabbit α-human β2GP1 antibody to generate smaller ICs (Figure 4A). IgG-containing α-β2GP1 ICs induced a 2.5- ± 0.2-fold increase in FXa activity, almost identical to the effect of HIT ICs. The induction of FXa by APS ICs was inhibited by 67.9% ± 3.3% (P < .0001) by SYNT001 (Figure 4A). Likewise, SYNT001 inhibited induction of TF by ICs formed with an affinity-purified IgG autoantibody from a patient with APS by 38.8% ± 6.7%.
![Induction of TF by APS and WAHA ICs. (A) Induction of TF by β2GP1 α-β2GP1 ICs. THP-1 cells were incubated for 3 hours with ICs formed between β2GP1 and IgG rabbit α-β2GP1 antibody (Ab) in the absence or presence of α−FcRn Ab or with αβ2GP1 Ab alone. TF activity was determined as in Figure 1A. Data (mean ± standard error of the mean [SEM]) from 4 independent experiments is shown. ***P < .0001 by 1-way ANOVA with Sidak’s multiple comparison test. (B) Induction of TF by IgG-coated RBCs and inhibition by α-FcRn Ab. Rh(D)+ or Rh(D)− RBCs were incubated with 100 ng/mL of mAb α-D IgG (BRAD-3) or α-D IgM (MAD-2). The RBCs were washed and added to THP-1 cells for 3 hours in the absence or presence of 200 µg/mL of α−FcRn Ab or an IgG4 control (Cont), and TF activity was measured as described in Figure 1A. Results from 2 to 5 independent experiments are shown (mean ± SEM). ***P < .0001, §P = .0003, and ¶P < .427 by 1-way ANOVA with Fisher’s least significant difference test.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/135/23/10.1182_blood.2019001133/2/m_bloodbld2019001133f4.png?Expires=1767721401&Signature=lXxILvEY6JK00LDasDtYF6ZOKUc8ZmJkDv-wh15DTQcvbkFcpvqn2k5MsLRblD~DuKCnyiZnuFP~hl09WsSEvXnGiKSVIGvnZWYXxcwahjEwXhgD0wp2PlFh-xypRFPna-UHuCvxqJjCGCipfmeQnN6C95kfrX04i9HJfTjupfVQ1gkdnJYbREFPmv4SZSicqXvJJhc05fijrDIscq8~RE51Habto0UuAOXoyAq65~lDRx4rINvukhYCaRXrPxcgklBit3DFKLpfeOa~qYbfQVZf~qsjjhMyGI9-LzXawwklrA6IeKtVGstXR6UcvQV7jE8p~uWZOP~EX~a3bhy8ww__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Induction of TF by APS and WAHA ICs. (A) Induction of TF by β2GP1 α-β2GP1 ICs. THP-1 cells were incubated for 3 hours with ICs formed between β2GP1 and IgG rabbit α-β2GP1 antibody (Ab) in the absence or presence of α−FcRn Ab or with αβ2GP1 Ab alone. TF activity was determined as in Figure 1A. Data (mean ± standard error of the mean [SEM]) from 4 independent experiments is shown. ***P < .0001 by 1-way ANOVA with Sidak’s multiple comparison test. (B) Induction of TF by IgG-coated RBCs and inhibition by α-FcRn Ab. Rh(D)+ or Rh(D)− RBCs were incubated with 100 ng/mL of mAb α-D IgG (BRAD-3) or α-D IgM (MAD-2). The RBCs were washed and added to THP-1 cells for 3 hours in the absence or presence of 200 µg/mL of α−FcRn Ab or an IgG4 control (Cont), and TF activity was measured as described in Figure 1A. Results from 2 to 5 independent experiments are shown (mean ± SEM). ***P < .0001, §P = .0003, and ¶P < .427 by 1-way ANOVA with Fisher’s least significant difference test.
Induction of TF by APS and WAHA ICs. (A) Induction of TF by β2GP1 α-β2GP1 ICs. THP-1 cells were incubated for 3 hours with ICs formed between β2GP1 and IgG rabbit α-β2GP1 antibody (Ab) in the absence or presence of α−FcRn Ab or with αβ2GP1 Ab alone. TF activity was determined as in Figure 1A. Data (mean ± standard error of the mean [SEM]) from 4 independent experiments is shown. ***P < .0001 by 1-way ANOVA with Sidak’s multiple comparison test. (B) Induction of TF by IgG-coated RBCs and inhibition by α-FcRn Ab. Rh(D)+ or Rh(D)− RBCs were incubated with 100 ng/mL of mAb α-D IgG (BRAD-3) or α-D IgM (MAD-2). The RBCs were washed and added to THP-1 cells for 3 hours in the absence or presence of 200 µg/mL of α−FcRn Ab or an IgG4 control (Cont), and TF activity was measured as described in Figure 1A. Results from 2 to 5 independent experiments are shown (mean ± SEM). ***P < .0001, §P = .0003, and ¶P < .427 by 1-way ANOVA with Fisher’s least significant difference test.
To examine whether the induction of TF by IgG ICs can be extended to particulates that are opsonized by IgG antibodies, THP-1 cells were incubated with human Rh(D)-expressing RBCs sensitized with an IgG mAb α-Rh(D) (BRAD-3). RBCs coated with BRAD-3 for 30 minutes increased the initial rate of FXa expression 3.1- ± 0.2-fold (Figure 4B). FXa was not generated by RBCs alone, by RBCs coated with an IgM α-Rh(D) mAb (MAD-2), or by Rh(D)− cells preincubated with BRAD-3, affirming the requirement for exposure to IgG-coated RBCs. SYNT001 inhibited FXa generation by BRAD-3 IgG–coated RBCs by 74.4% ± 11.1% vs 28.3% ± 9.0% by control IgG4 based on initial velocities (P = .0293 by Student t test; data not shown) and by 50% ± 9% (P = .0004) based on fold increase relative to IgG4 (Figure 4B). Similar results were obtained using human polyclonal IgG α-Rh(D) (RhoGAM). Human IgG antibody–coated RBCs increased FXa activity 2.4- ± 0.1-fold (P = 1 × 10e−5; n = 5), and FXa generation was inhibited by 85.5% ± 1.1% with 200 µg/mL of SYNT001 (P = .0002; supplemental Figure 4).
Involvement of FcRn in vivo
We have previously shown that infusion of KKO induces a prothrombotic state in hPF4+/FcγRIIa+ transgenic mice,11,42,45,48 characterized by platelet adhesion and fibrin deposition in cremaster muscle arterioles after laser injury. Therefore, we studied the effect of antibody-mediated blockade of FcRn on the KKO-induced prothrombotic state in vivo. Injection of a mouse α-human FcRn mAb that cross-reacts with mouse FcRn (DVN24)38 did not significantly affect platelet deposition (Figure 5A) but efficiently blocked fibrin accumulation in hPF4+/FcγRIIa+ mice (Figure 5B-C). Monocytes from HIT mice preinjected with DVN24 generated significantly less FXa after stimulation by HIT ICs than monocytes from mice given isotype control (maximal velocity, 15.1 ± 4.0 vs 10.5 ± 3.8 mOD per minute; n = 9 mice per group; P = .004).
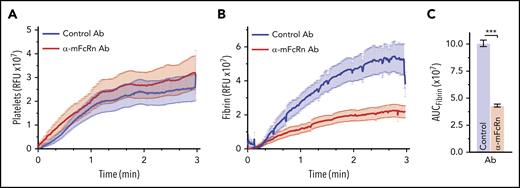
Inhibition of fibrin deposition by α-FcRn antibody in a murine model of HIT. Widefield cremaster muscle arteriole laser injuries were performed in human PF4+/FcγRIIa+ mice pretreated with 200 µg per mouse of α-mouse FcRn (α-mFcRn) antibody (Ab; DVN24) or IgG2a isotype control. Three mice from each arm were studied, and each mouse had 5 to 8 injuries. Accumulations of platelets (A) and fibrin (B) in relative fluorescence units (RFUs) over time are shown. (C) Total accumulation was determined by calculating the area under the curve (AUC) for each injury. ***P < .0001 by Student t test with Welch’s correction.
Inhibition of fibrin deposition by α-FcRn antibody in a murine model of HIT. Widefield cremaster muscle arteriole laser injuries were performed in human PF4+/FcγRIIa+ mice pretreated with 200 µg per mouse of α-mouse FcRn (α-mFcRn) antibody (Ab; DVN24) or IgG2a isotype control. Three mice from each arm were studied, and each mouse had 5 to 8 injuries. Accumulations of platelets (A) and fibrin (B) in relative fluorescence units (RFUs) over time are shown. (C) Total accumulation was determined by calculating the area under the curve (AUC) for each injury. ***P < .0001 by Student t test with Welch’s correction.
Discussion
This study demonstrates that FcRn contributes to the induction of TF activity on monocytic cells exposed to soluble IgG-containing ICs generated from antigens that are involved in the pathogenesis of HIT and APS and by particulate complexes composed of IgG antibody–coated RBCs that cause WAHA. First, we find that a humanized mAb that blocks IgG interactions with FcRn, SYNT001, interferes with TF-dependent FXa generation in response to each of these ICs. Second, TF-dependent FXa activity is induced by engineered ICs containing a wild-type IgG Fc that binds to FcγR and FcRn but not those formed by antibodies with an Fc containing a mutation that specifically disables its engagement with FcRn. Third, fibrin accumulation is inhibited by an α-mouse FcRn mAb after vascular injury in a murine model of HIT in which TF is generated in monocytes.9,41,45 Together, these studies reveal that induction of TF activity requires not only FcγRIIa, as we have previously shown,42,45 but also FcRn, as we describe here.
The mechanism by which FcRn participates in the induction of TF-dependent FXa activity by IgG-containing ICs will require additional studies. One possibility is that the signal transducing activity leading to activation of TF is initiated within the cell where acidic endosomes reside and contain the milieu necessary for IgG-containing IC binding to FcRn. Soluble IgG-containing ICs bound to FcγRs, internalized by clathrin-dependent endocytosis, and particulate complexes, internalized by phagocytosis, are presumed to develop prolonged interactions with FcRn at the acidic pH found in early sorting and especially later endosomes.17,20 FcRn may also modulate the stability of ICs in the late endosomes where β2GP1 accumulates22,49 and thereby orchestrate signal transduction alone16,50 and/or in collaboration with TLR-4, TLR-2, or other receptors51,52 involved in the induction of TF activity.13,15,21 Importantly, phagocytosis of IgG-opsonized particles by neutrophils depends on FcRn,53 consistent with the role of this receptor in determining innate and adaptive immune responses by hematopoietic cells and its ability to regulate intracellular signaling in response to ICs,54 which, as we show here, likely extends to the induction of TF activity.
Our data demonstrate that the induction of TF-dependent FXa activity by monocytic cells in response to ICs depends not only on FcγRIIa, as previously described,9-11 but also on FcRn, as shown here. Because HUVECs express FcRn but not FcγRs and did not increase TF-dependent FXa activity when exposed to IgG-containing ICs, our study suggests that both types of receptors are required. Consistent with this, soon after exposure of THP-1 cells to IgG-containing ICs, we observed a redistribution of FcRn into clusters that contained FcγRIIa. Additional research is needed to understand how FcRn and FcγRIIa may cooperate in t3his process. However, the finding that FcγRIIa-mediated induction of FXa activity involves FcRn is consistent with other FcγR-dependent processes that require FcRn, such as the induction of innate immune cytokine production,50 antigen presentation by dendritic cells,16 and phagocytosis of IgG-containing ICs by neutrophils.53
α-FcRn antibodies such as SYNT001 are in clinical trials in several IgG antibody–mediated disorders, including WAHA, immune thrombocytopenia, and pemphigus, with the goal of lowering plasma levels of IgG autoantibodies and IgG-containing ICs in addition to inhibiting the ability of IgG-containing ICs to induce inflammatory responses associated with innate and adaptive immune pathways.17 The results of this study suggest that α-FcRn therapies may also lower the risk of thromboembolic complications caused by IgG-containing ICs in these and other immune/inflammatory disorders associated with an increased risk of thrombosis.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Vincent Hayes for his invaluable technical assistance.
This work was supported by National Institutes of Health (NIH) National Heart, Lung, and Blood Institute grants ROHL142122, R01 HL139448, and R01 HL128895 (D.B.C.), R01 HL141462 (V.S.), R01 HL068835 (A.E.M.), R01 HL125422 and P01 HL139420 (S.K.), R01 HL123098 (K.R.M.), and NIH National Institute of Diabetes and Digestive and Kidney Diseases grant R01 DK053056 (R.S.B.); and by sponsored research support from Syntimmune/Alexion.
Authorship
Contribution: D.B.C., L.J.B., J.J.H., M. Pyzik, and R.S.B. designed the research; M. Poncz, A.S., and L.R. designed and performed the in vivo experiments; A.E.M. performed the studies involving TFPI; K.R.M. characterized β2GPI protein and antibodies; S.K. designed and helped to interpret measurements of FXa; M. Pyzik, A.H.R., S.Z., and V.S. designed, analyzed, and interpreted all in vitro studies; M.A.K. and G.Z. designed, performed, and analyzed experiments involving messenger RNA expression; and D.B.C., M. Pyzik, L.R., and R.S.B. wrote the manuscript.
Conflict-of-interest disclosure: L.J.B. was an employee of Syntimmune, Inc. R.S.B. served as consultant to Syntimmune, Inc., and had equity interests in Syntimmune, Inc., a company developing therapeutic agents to target FcRn. Syntimmune, Inc. is now a wholly owned subsidiary of Alexion Pharmaceuticals, Inc., after its acquisition by Alexion. The remaining authors declare no competing financial interests.
Correspondence: Douglas B. Cines, 513A Stellar-Chance, 422 Curie Blvd, Philadelphia, PA 19104; e-mail: dcines@pennmedicine.upenn.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal