

Key Points
Age had a significant association with OS, with superior OS observed in patients <5 years of age at the time of HCT.
Donor myeloid engraftment <50% was associated with inferior platelet recovery after HCT.

Abstract
Wiskott-Aldrich syndrome (WAS) is an X-linked disease caused by mutations in the WAS gene, leading to thrombocytopenia, eczema, recurrent infections, autoimmune disease, and malignancy. Hematopoietic cell transplantation (HCT) is the primary curative approach, with the goal of correcting the underlying immunodeficiency and thrombocytopenia. HCT outcomes have improved over time, particularly for patients with HLA-matched sibling and unrelated donors. We report the outcomes of 129 patients with WAS who underwent HCT at 29 Primary Immune Deficiency Treatment Consortium centers from 2005 through 2015. Median age at HCT was 1.2 years. Most patients (65%) received myeloablative busulfan-based conditioning. With a median follow-up of 4.5 years, the 5-year overall survival (OS) was 91%. Superior 5-year OS was observed in patients <5 vs ≥5 years of age at the time of HCT (94% vs 66%; overall P = .0008). OS was excellent regardless of donor type, even in cord blood recipients (90%). Conditioning intensity did not affect OS, but was associated with donor T-cell and myeloid engraftment after HCT. Specifically, patients who received fludarabine/melphalan-based reduced-intensity regimens were more likely to have donor myeloid chimerism <50% early after HCT. In addition, higher platelet counts were observed among recipients who achieved full (>95%) vs low-level (5%-49%) donor myeloid engraftment. In summary, HCT outcomes for WAS have improved since 2005, compared with prior reports. HCT at a younger age continues to be associated with superior outcomes supporting the recommendation for early HCT. High-level donor myeloid engraftment is important for platelet reconstitution after either myeloablative or busulfan-containing reduced intensity conditioning. (This trial was registered at www.clinicaltrials.gov as #NCT02064933.)
Introduction
Wiskott Aldrich syndrome (WAS) is an X-linked disorder with an estimated incidence of 1 in 100 000 live male births,1 caused by hemizygous mutations in the WAS gene and characterized by microthrombocytopenia, eczema, progressive T- and B-cell immunodeficiency, and increased risk for autoimmune disease and malignancy.2 The genetic defect, located at Xp11.22,3 was first identified in 1994.4 The WAS gene encodes the WAS protein (WASP), which is expressed only in hematopoietic cells, except red blood cells,4,5 and is important for transduction of signals from receptors on the cell surface to the actin cytoskeleton. WASP plays a crucial role in actin cytoskeleton remodeling6 and in the formation and stability of the immunologic synapse between T and B cells, and between natural killer and regulatory T cells with their target cells.7 Patients with WAS have chronic morbidities and a decreased life expectancy that is caused by an increased risk of life-threatening infections, autoimmune complications, bleeding, and malignancies.2,8-10
In the modern era, the median survival of patients with WAS has increased from 6.5 years to early adulthood,2,9 because of improved prophylactic antimicrobials, immunoglobulin supplementation, symptomatic management of eczema, and autoimmune complications, and implementation of bleeding precautions. Allogeneic hematopoietic cell transplantation (HCT), which has the potential to provide long-term cure, is the primary definitive treatment approach for patients with WAS. Successful HCT for WAS was first reported in 1968,11 and several large retrospective analyses are now available.12-17 These studies demonstrated that patients who received an HLA-identical sibling graft and underwent HCT at a younger age (<5 years), had improved survival. Of note, <50% myeloid donor chimerism was found to be associated with an increased risk of persistent thrombocytopenia, and mixed donor chimerism was associated with autoimmune complications after HCT.14,16
We report on 129 patients with WAS who underwent HCT between 2005 and 2015 at 29 US and Canadian centers of the Primary Immune Deficiency Treatment Consortium (PIDTC). The primary objective of this study was to evaluate survival after HCT and the effect of various factors, including age at HCT, WAS expression, WAS score, donor type, hematopoietic cell source, and conditioning regimen intensity on survival. We also examined the effect of donor chimerism on platelet recovery and incidence of post-HCT autoimmune complications.
Materials and methods
Study participants
PIDTC protocol 690418 is a multicenter observational study evaluating outcomes of patients with WAS who had undergone HCT since 1990. The institutional review boards of all participating PIDTC centers approved the study and approved a waiver of consent for all patients on this retrospective study. We report the outcomes of a subset of patients from the retrospective arm of the study who underwent HCT between 2005 and 2015. After a central review, male patients were deemed eligible if they met one of the following diagnostic criteria: (1) thrombocytopenia defined as a platelet count <100 000/μL and either (a) a molecular diagnosis of WAS or (b) absent or reduced WASP expression; (2) chronic thrombocytopenia, defined as a platelet count <100 000/μL on 2 evaluations separated by at least 3 months and a positive family history of WAS; or (3) chronic thrombocytopenia and a low mean platelet volume (below normal for age) and either (a) recurrent and/or severe infections requiring treatment and/or eczema or (b) lack of antibody response to polysaccharide antigens and/or low IgM. Additional clinical and laboratory data were also taken into consideration by the PIDTC-WAS Eligibility Review Committee when information was limited.
Pre-HCT patient characteristics are listed in Table 1. Infections and autoimmune complications were also recorded. We defined autoimmune thrombocytopenia (vs reduced platelet production) as any of the following: (1) low platelet counts that did not increase appropriately after platelet transfusions; (2) severe bleeding that did not resolve following platelet transfusions; (3) significant platelet count response to steroids, rituximab, or immunoglobulin infusion; (4) the presence of platelet autoantibodies; or (5) the presence of increased megakaryocytes in the bone marrow.
Characteristics of patients before HCT
| Characteristic | Data (N = 129) |
|---|---|
| Year of HCT | |
| 2005-2010 | 79 (61) |
| 2011-2015 | 50 (39) |
| Age at HCT, y | |
| <1 | 53 (41) |
| 1-1.99 | 40 (31) |
| 2-4.99 | 24 (19) |
| ≥5 | 12 (9) |
| Lansky/Karnofsky | |
| Median (range) | 100 (60-100) |
| Family history of WAS | |
| Yes | 48 (37) |
| No | 69 (54) |
| Unknown | 12 (9) |
| History of eczema before HCT | |
| Yes | 111 (86) |
| No | 15 (12) |
| Unknown | 3 (2) |
| History of chronic thrombocytopenia before HCT* | |
| Yes | 109 (85) |
| No | 13 (10) |
| Unknown | 7 (5) |
| History of splenectomy before HCT | |
| Yes | 8 (6) |
| No | 121 (94) |
| WAS mutations identified | |
| Nonsense | 30 (23) |
| Frameshift | 34 (26) |
| Deletion | 21 (16) |
| Insertion | 12 (9) |
| Deletion/insertion | 1 (1) |
| Missense | 30 (23) |
| Splice site | 20 (15) |
| Invariant | 13 (10) |
| Variant | 7 (5) |
| In-frame insertion | 2 (2) |
| Large deletion | 5 (4) |
| Unknown | 8 (6) |
| WASP expression by flow cytometry or predicted | |
| Absent | 83 (64) |
| Reduced | 42 (33) |
| Normal | 2 (1.5) |
| Unknown | 2 (1.5) |
| History of recurrent or severe infections before HCT | |
| Yes | 64 (50) |
| No | 65 (50) |
| History of autoimmune disease before HCT/present at time of conditioning | |
| Yes | 32 (25)/26 (20) |
| Thrombocytopenia | 19 (15)/17 (13) |
| Hemolytic anemia | 10 (8)/6 (5) |
| Neutropenia | 7 (5)/5 (4) |
| Vasculitis | 3 (2)/1 (1) |
| Inflammatory bowel disease | 2 (2)/2 (2) |
| Arthritis | 1 (1)/1 (1) |
| Nephritis | 1 (1)/1 (1) |
| Alopecia | 1 (1)/1 (1) |
| No | 91 (70) |
| Unknown | 6 (5) |
| Characteristic | Data (N = 129) |
|---|---|
| Year of HCT | |
| 2005-2010 | 79 (61) |
| 2011-2015 | 50 (39) |
| Age at HCT, y | |
| <1 | 53 (41) |
| 1-1.99 | 40 (31) |
| 2-4.99 | 24 (19) |
| ≥5 | 12 (9) |
| Lansky/Karnofsky | |
| Median (range) | 100 (60-100) |
| Family history of WAS | |
| Yes | 48 (37) |
| No | 69 (54) |
| Unknown | 12 (9) |
| History of eczema before HCT | |
| Yes | 111 (86) |
| No | 15 (12) |
| Unknown | 3 (2) |
| History of chronic thrombocytopenia before HCT* | |
| Yes | 109 (85) |
| No | 13 (10) |
| Unknown | 7 (5) |
| History of splenectomy before HCT | |
| Yes | 8 (6) |
| No | 121 (94) |
| WAS mutations identified | |
| Nonsense | 30 (23) |
| Frameshift | 34 (26) |
| Deletion | 21 (16) |
| Insertion | 12 (9) |
| Deletion/insertion | 1 (1) |
| Missense | 30 (23) |
| Splice site | 20 (15) |
| Invariant | 13 (10) |
| Variant | 7 (5) |
| In-frame insertion | 2 (2) |
| Large deletion | 5 (4) |
| Unknown | 8 (6) |
| WASP expression by flow cytometry or predicted | |
| Absent | 83 (64) |
| Reduced | 42 (33) |
| Normal | 2 (1.5) |
| Unknown | 2 (1.5) |
| History of recurrent or severe infections before HCT | |
| Yes | 64 (50) |
| No | 65 (50) |
| History of autoimmune disease before HCT/present at time of conditioning | |
| Yes | 32 (25)/26 (20) |
| Thrombocytopenia | 19 (15)/17 (13) |
| Hemolytic anemia | 10 (8)/6 (5) |
| Neutropenia | 7 (5)/5 (4) |
| Vasculitis | 3 (2)/1 (1) |
| Inflammatory bowel disease | 2 (2)/2 (2) |
| Arthritis | 1 (1)/1 (1) |
| Nephritis | 1 (1)/1 (1) |
| Alopecia | 1 (1)/1 (1) |
| No | 91 (70) |
| Unknown | 6 (5) |
Unless otherwise specified, data are the number of recipients (percentage of total study group).
Chronic thrombocytopenia is defined as a platelet count <100 000/μL on 2 evaluations separated by at least 3 months.
Mutation analysis, WASP expression, and WAS score
Information regarding the WAS gene8,19 and WASP expression5,8,20 was recorded and reviewed centrally by the PIDTC-WAS Eligibility Review Committee to confirm the diagnosis of WAS. The WAS severity scoring system9 was used to differentiate patients with mild from those with more severe clinical phenotypes. Subjects with a “mild” WAS phenotype were characterized as having thrombocytopenia with absent or isolated and intermittent eczematous lesions that responded promptly to local therapy and absence of recurrent or chronic infections (WAS score 1 or 2; often referred to as X-linked thrombocytopenia). Patients with classic WAS had extensive and difficult-to-treat eczema or had recurrent infections requiring frequent antimicrobial agents (score 3 or 4). Patients who developed autoimmunity or malignancy received a score of 5.9 WAS scores were based predominately on the severity of eczema and infections, as well as the presence of autoimmunity, because information describing the pre-HCT immune status was limited. The WAS score is less reliable in patients <2 years of age8 ; therefore, WASP expression was also taken into consideration for this age group.
HCT
Age and Lansky/Karnofsky score at HCT, conditioning regimen, donor type, hematopoietic cell source, including degree of HLA match, and graft-versus-host-disease (GVHD) prophylaxis, including T-cell depletion, were recorded. Bone marrow and peripheral blood stem cell (PBSC) grafts were considered fully matched (10 of 10) if matched at HLA-A, -B, -C, -DRB1, and DQB1 at the allele level (unrelated donors) or antigen level (sibling donors). Cord blood (CB) grafts were considered matched (6 of 6) if matched at HLA-A and -B (antigen level) and -DRB1 (allele level). The conditioning regimens were categorized as either myeloablative or reduced intensity, based on the following definitions.21-25 Myeloablative conditioning (MAC) was defined as regimens that contained one of the following: (1) busulfan area under the curve (AUC) ≥50 mg × h/L combined with cyclophosphamide or melphalan; (2) busulfan AUC ≥80 mg × h/L combined with fludarabine21 ; or (3) if busulfan AUC data were unavailable, busulfan at a total dose of ≥12 mg/kg combined with cyclophosphamide.22,23 Reduced-intensity conditioning (RIC) included one of the following: (1) a busulfan cumulative AUC <50 mg × h/L combined with cyclophosphamide; or (2) a busulfan cumulative AUC <80 mg × h/L combined with fludarabine; or (3) if busulfan AUC data were unavailable, busulfan at a total dose of ≤8 mg/kg.24 Other RIC regimens consisted primarily of fludarabine and melphalan, with or without thiotepa.
The results of lineage-specific donor chimerism in whole-blood myeloid cells (CD33+, CD15+, or CD14+), CD3+ T cells, and CD19+ B cells were also recorded. Donor chimerism was defined as full (>95%), high-level (50%-95%), or low-level mixed (5%-49%) in the lineage measured. Graft failure/rejection was defined as <5% donor cells in CD3 T-cell and/or myeloid lineages by 1 year after HCT. When more than 1 lineage-specific chimerism value was available, the chimerism value closest to the time point of interest was used. Information regarding acute grade 2-4, 3-4, and chronic GVHD26 ; post-HCT autoimmune complications, and cause of death was also recorded. Data were stored in the Data Management Coordinating Center of the Rare Diseases Clinical Research Network.
Statistical analysis
Descriptive statistics, such as medians and ranges for continuous variables and counts and percentages for categorical variables, were used to summarize patient demographic, disease, and treatment-related variables. The Wilcoxon rank-sum or Kruskal-Wallis test was used to compare continuous variables between groups. Categorical variables were compared by using the χ2 test or Fisher’s exact test where needed. The Kaplan-Meier method was used to estimate the probability of OS. Death from any cause was considered as an event, and surviving patients were censored at last follow-up. Log-rank test was used to compare OS probabilities between different groups of patients. The probabilities of hematopoietic recovery and acute and chronic GVHD were calculated using the cumulative incidence function estimator, with death as a competing risk. The 95% confidence interval (CI) was estimated by using log-log transformation. All P-values are 2-sided, and analyses were performed with SAS software, version 9.4 (SAS Institute, Cary, NC). For patients who received a second HCT or boost, chimerism and platelet data were analyzed up to the time when these events occurred.
Results
Baseline patient characteristics
From January 2005 through December 2015, 129 patients who underwent HCT for WAS at 29 PIDTC centers were enrolled in the study. Patient and HCT characteristics are shown in Tables 1 and 2. Most patients underwent HCT before the age of 2 years (n = 93), with a median age at HCT of 1.2 (range, 0.2-21.7) years (Figure 1). A pre-HCT WAS score (range, 1-5) was determined for each patient (Table 3). Fifty-three patients (41%) had a WAS score of 1 to 2, the majority of whom were <2 years of age at time of HCT (n = 43). Forty-three patients (33%) had a WAS score of 3 to 4 indicating a severe WAS phenotype, and 33 (26%) exhibited the most severe phenotype with a score of 5. Data on the WAS gene mutations were available for 121 patients (94%). Most patients had a severe mutation (nonsense, frameshift, invariant splice-site, or large deletions; n = 82), compared with patients with less severe mutations (missense, variant splice-site, and in-frame insertion; n = 39). Of the 129 patients, 127 had data available on WASP expression (n = 55) or had WASP expression predicted based on the WAS mutation (n = 72). Of the 127 evaluable patients, 83 had absent, 42 had reduced, and 2 had normal WASP expression.
Donor and transplant characteristics
| Characteristics | Data (N = 129) |
|---|---|
| Donor matching | |
| HLA-matched sibling | 22 (17) |
| HLA-mismatched related* | 2 (2) |
| HLA-matched unrelated (10 of 10) | 41 (32) |
| HLA-mismatched unrelated (≤ 9 of 10) | 21 (16) |
| Unrelated CB | 39 (30) |
| 6 of 6 | 13 (10) |
| 5 of 6 | 20 (15) |
| 4 of 6 | 6 (5) |
| Unrelated donor, match unknown | 4 (3) |
| Hematopoietic stem cell source | |
| Bone marrow† | 80 (62) |
| PBSC | 10 (8) |
| CB | 39 (30) |
| Conditioning regimen‡ | |
| Myeloablative | 88 (68) |
| Busulfan/cyclophosphamide | 80 (62) |
| Busulfan/fludarabine | 3 (2) |
| Busulfan/melphalan | 1 (1) |
| MAC (other) | 4 (3) |
| Reduced intensity | 39 (30) |
| Busulfan/cyclophosphamide | 8 (6) |
| Busulfan/fludarabine | 17 (13) |
| RIC (other) | 14 (11) |
| Fludarabine/melphalan ± thiotepa | 12 (9) |
| Other | 2 (2) |
| Unknown intensity | 2 (2) |
| Busulfan/fludarabine | 2 (2) |
| Serotherapy | |
| Yes | 123 (95) |
| Rabbit or horse ATG | 99 (76) |
| Alemtuzumab | 24 (19) |
| No | 6 (5) |
| GVHD prophylaxis | |
| CNI alone | 2 (2) |
| CNI+MTX | 41 (32) |
| CNI+MMF | 21 (16) |
| CNI+steroids | 49 (38) |
| CNI+steroids+MTX | 11 (8) |
| CNI+steroids+MMF | 1 (1) |
| None | 4 (3)§ |
| T-cell depletion | |
| Yes | 5 (4) |
| No | 124 (96) |
| Characteristics | Data (N = 129) |
|---|---|
| Donor matching | |
| HLA-matched sibling | 22 (17) |
| HLA-mismatched related* | 2 (2) |
| HLA-matched unrelated (10 of 10) | 41 (32) |
| HLA-mismatched unrelated (≤ 9 of 10) | 21 (16) |
| Unrelated CB | 39 (30) |
| 6 of 6 | 13 (10) |
| 5 of 6 | 20 (15) |
| 4 of 6 | 6 (5) |
| Unrelated donor, match unknown | 4 (3) |
| Hematopoietic stem cell source | |
| Bone marrow† | 80 (62) |
| PBSC | 10 (8) |
| CB | 39 (30) |
| Conditioning regimen‡ | |
| Myeloablative | 88 (68) |
| Busulfan/cyclophosphamide | 80 (62) |
| Busulfan/fludarabine | 3 (2) |
| Busulfan/melphalan | 1 (1) |
| MAC (other) | 4 (3) |
| Reduced intensity | 39 (30) |
| Busulfan/cyclophosphamide | 8 (6) |
| Busulfan/fludarabine | 17 (13) |
| RIC (other) | 14 (11) |
| Fludarabine/melphalan ± thiotepa | 12 (9) |
| Other | 2 (2) |
| Unknown intensity | 2 (2) |
| Busulfan/fludarabine | 2 (2) |
| Serotherapy | |
| Yes | 123 (95) |
| Rabbit or horse ATG | 99 (76) |
| Alemtuzumab | 24 (19) |
| No | 6 (5) |
| GVHD prophylaxis | |
| CNI alone | 2 (2) |
| CNI+MTX | 41 (32) |
| CNI+MMF | 21 (16) |
| CNI+steroids | 49 (38) |
| CNI+steroids+MTX | 11 (8) |
| CNI+steroids+MMF | 1 (1) |
| None | 4 (3)§ |
| T-cell depletion | |
| Yes | 5 (4) |
| No | 124 (96) |
Data are number of recipients (percentage of total study group).
ATG, anti-thymocyte globulin.
Two patients received HLA-mismatched related grafts that included a 5- of 8-HLA–mismatched relative and a 9- of 10-HLA–matched sibling graft.
Two patients received bone marrow plus CB from their siblings.
Of the 108 patients who received busulfan/cyclophosphamide or busulfan/fludarabine, AUC data were available in 89 patients and were used for classification of MAC (n = 64) and RIC (n = 25). Two additional patients who received busulfan/fludarabine could not be classified because of lack of information and were excluded from analyses comparing MAC and RIC.
Four patients who received TCD grafts received no additional immune suppression for GVHD prophylaxis.
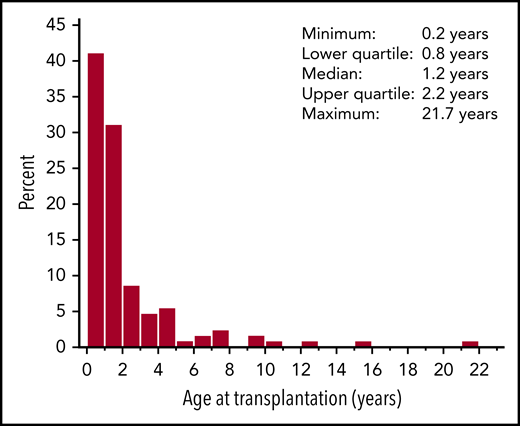
Age at HCT. Histogram shows percentage of subjects at ages 0 to 22 years at the time of HCT.
Age at HCT. Histogram shows percentage of subjects at ages 0 to 22 years at the time of HCT.
WAS score by age
| WAS Score | Age, y | |||
|---|---|---|---|---|
| <1 (n = 53) | 1-1.99 (n = 40) | 2-4.99 (n = 24) | ≥5 (n = 12) | |
| 1–2 (n = 53) | 29 (55) | 14 (35) | 6 (25) | 4 (33) |
| 3–4 (n = 43) | 13 (24) | 15 (37.5) | 13 (54) | 2 (17) |
| 5 (n = 33) | 11 (21) | 11 (27.5) | 5 (21) | 6 (50) |
| WAS Score | Age, y | |||
|---|---|---|---|---|
| <1 (n = 53) | 1-1.99 (n = 40) | 2-4.99 (n = 24) | ≥5 (n = 12) | |
| 1–2 (n = 53) | 29 (55) | 14 (35) | 6 (25) | 4 (33) |
| 3–4 (n = 43) | 13 (24) | 15 (37.5) | 13 (54) | 2 (17) |
| 5 (n = 33) | 11 (21) | 11 (27.5) | 5 (21) | 6 (50) |
Data are expressed as the number of recipients (percentage of total in age subgroup).
Sixty-four patients had a history of 1 or more recurrent or severe infection requiring treatment pre-HCT. In addition, 32 patients (25%) had a history of 1 or more autoimmune problems before HCT, of whom 26 had autoimmunity at the time of conditioning. Cytopenias were the most common autoimmune problem before HCT (thrombocytopenia [n = 19], hemolytic anemia [n = 10], or neutropenia [n = 7]), most of which persisted at the time of conditioning. One patient had a history of malignancy (cutaneous T-cell lymphoma) before HCT.
HCT characteristics
Information regarding conditioning regimen and intensity was available for 127 patients. Most patients received MAC (n = 88), with busulfan-based conditioning (n = 84) being the most common (Table 2). Thirty-nine patients received RIC, with busulfan-based conditioning (n = 25) again being the most common. Of note, for most patients who received busulfan/cyclophosphamide or busulfan/fludarabine, AUC data were available and used for classification of MAC vs RIC (n = 89). Approximately one-third of the recipients of bone marrow and CB received RIC (Table 4). Most patients also received serotherapy (n = 123) with rabbit or horse anti-thymocyte globulin (n = 99) or alemtuzumab (n = 24). GVHD prophylaxis consisted primarily of a calcineurin inhibitor (CNI, cyclosporine, or tacrolimus) in combination with methotrexate (MTX; n = 41), mycophenolate mofetil (MMF; n = 21), or steroids (n = 49). Four patients who received T-cell–depleted (TCD) grafts did not receive additional immune suppression for GVHD prophylaxis.
Conditioning intensity and hematopoietic cell source
| Hematopoietic cell source | MAC | RIC | Missing | Total |
|---|---|---|---|---|
| BM or BM+CB | 55 (69) | 24 (30) | 1 (1) | 80 |
| PBSC | 5 (50) | 4 (40) | 1 (10) | 10 |
| Unrelated CB | 28 (72) | 11 (28) | 0 | 39 |
| Hematopoietic cell source | MAC | RIC | Missing | Total |
|---|---|---|---|---|
| BM or BM+CB | 55 (69) | 24 (30) | 1 (1) | 80 |
| PBSC | 5 (50) | 4 (40) | 1 (10) | 10 |
| Unrelated CB | 28 (72) | 11 (28) | 0 | 39 |
Data are expressed as the number of recipients (percentage of total in hematopoieitc cell source subgroup).
BM, bone marrow.
The majority (n = 80) of patients received bone marrow grafts. Data on HLA matching were available for 125 patients. Patients received HLA-matched sibling (n = 22), mismatched related (n = 2), matched unrelated (n = 41), or mismatched unrelated (n = 21) grafts. In addition, 39 patients received unrelated CB grafts that matched at 6 of 6 (n = 13), 5 of 6 (n = 20), or 4 of 6 (n = 6) loci. There was no significant difference in the median age at HCT or in median WAS score based on donor type (HLA-matched related, matched unrelated, mismatched unrelated, or CB; data not shown).
GVHD
Information regarding acute and chronic GVHD was available for 128 and 123 patients, respectively. The cumulative incidence of day 100 acute grades 2 to 4 and 3 to 4 and 1-year chronic GVHD were 27% (95% CI, 20%-35%), 15% (95% CI, 9%-22%), and 17% (95% CI, 11%-24%), respectively (Figure 2). There was no difference in the cumulative incidence of acute grades 2 to 4 and 3 to 4 and 1-year chronic GVHD in recipients of HLA-matched sibling, matched unrelated, and CB grafts (data not shown).
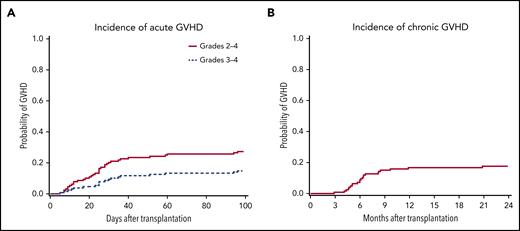
Probabilities of acute and chronic GVHD in patients with WAS after HCT. Shown is the cumulative incidence of acute grades 2 to 4 and 3 to 4 GVHD (A) (n = 128) up to 100 days after HCT and of chronic GVHD (B) (n = 123) up to 24 months.
Probabilities of acute and chronic GVHD in patients with WAS after HCT. Shown is the cumulative incidence of acute grades 2 to 4 and 3 to 4 GVHD (A) (n = 128) up to 100 days after HCT and of chronic GVHD (B) (n = 123) up to 24 months.
OS
At a median follow-up of 4.5 (range, 0.4-12.1) years, 117 patients were alive. The estimated OS at 5 years was 91% (95% CI, 85%-95%). Age at HCT was the only factor that had a significant association with OS, with superior OS in patients who were <5 years of age (n = 117) compared with those who were ≥5 years (n = 12) at the time of HCT (5-year OS: 94% [95% CI, 88%-97%] vs 66% [95% CI, 32%-86%], respectively [overall P = .0008; Figure 3A]). Excellent 5-year OS was seen among infants (<1 year, 94% [95% CI, 83%-98%]) young children (1-1.99 years, 95% [95% CI, 81%-99%], and children 2 to 4.99 years of age (92% [95% CI, 70%-98%]). Conditioning regimen intensity, donor type, hematopoietic cell source, HCT year, WAS score, and WASP expression were not significantly associated with OS (Figure 3B-D; the data for HCT year, WAS score, and WAS expression are not shown).
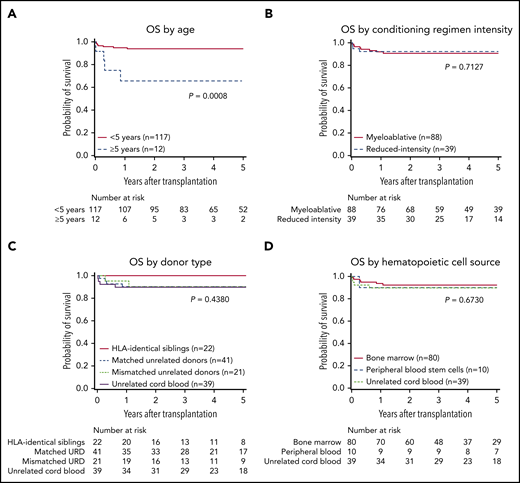
OS probabilities in 129 patients with WAS. Shown are the probabilities of OS by age (A), conditioning regimen intensity (B), donor type (C), and hematopoietic cell source (D). The P-values were obtained by log-rank test.
OS probabilities in 129 patients with WAS. Shown are the probabilities of OS by age (A), conditioning regimen intensity (B), donor type (C), and hematopoietic cell source (D). The P-values were obtained by log-rank test.
Sixteen patients developed 1 or more severe organ complications within the first 100 days after HCT. Five patients developed sinusoidal obstructive syndrome, 2 required dialysis, and 3 developed cardiac complications. In addition, 9 patients required mechanical ventilation because of infections (n = 4), pulmonary hemorrhage (n = 2; 1 in association with idiopathic pneumonia syndrome and SOS), aspiration (n = 1), recurrent seizures (n = 1), and intracranial hemorrhage (n = 1). Almost all patients received MAC busulfan/cyclophosphamide (n = 10) or RIC busulfan/cyclophosphamide or busulfan/fludarabine (n = 4). Twelve patients died, most (10 of 12) within the first year after HCT. Causes of death included sepsis (n = 3), GVHD with or without infection (n = 4), hemorrhage (n = 3; intracranial [n = 2] and pulmonary [n = 1]), and multiorgan failure (n = 2). None of the 8 patients who underwent a splenectomy before HCT had died at the time of last follow-up.
Engraftment and chimerism
Most patients evaluated had full donor (>95%) CD3 T-cell, CD19 B-cell, and myeloid chimerism at day 100, 6 months, 1 year, and 2 years after HCT (Figure 4). There was a low rate of graft rejection [<5% donor chimerism in CD3 T-cell and/or myeloid fractions; (n = 4)] by 1 year after HCT. However, 6 patients required a second HCT (n = 5) or marrow boost without conditioning (n = 1) due to graft rejection (n = 4) or continued thrombocytopenia in the setting of low-level donor myeloid engraftment (n = 2). Five of the 6 patients had received RIC conditioning followed by a bone marrow graft. All 6 patients were alive, and 5 had high-level or full donor engraftment and platelet counts >100 000/μL at their last follow-up. In addition, 1 patient who received 6 planned donor lymphocyte infusions following a CD34-selected, TCD, HLA-mismatched-related PBSC HCT, remained alive with high-level whole-blood chimerism at his last follow-up.
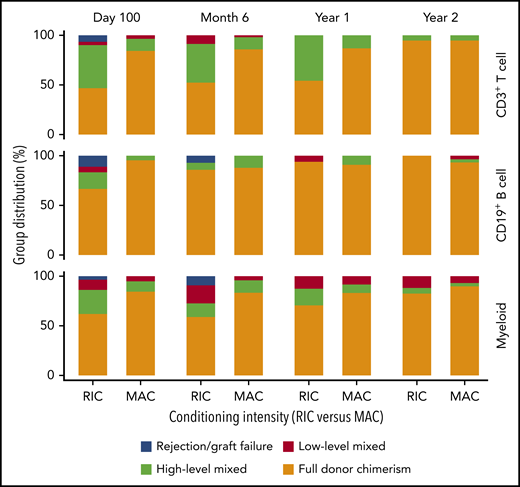
Donor CD3+ T-cell, CD19+ B-cell, and myeloid chimerism vs conditioning regimen intensity (RIC vs MAC) over the first 2 years after HCT. Donor chimerism: full, >95%; high-level, 50% to 95%; low-level, 5% to 49%; rejection, <5%. MAC resulted in a higher percentage of patients with full donor (>95%) CD3+ T-cell chimerism compared with RIC at day 100 (P < .001), 6 months (P = .006), and 1 year (P = .002) after HCT. This difference was not significant at 2 years after HCT. MAC resulted in a higher percentage of patients with full donor (>95%) B-cell chimerism at day 100 (P = .004) and myeloid chimerism at 6 months (P = .023), compared with RIC. The differences at all other time points were not significant.
Donor CD3+ T-cell, CD19+ B-cell, and myeloid chimerism vs conditioning regimen intensity (RIC vs MAC) over the first 2 years after HCT. Donor chimerism: full, >95%; high-level, 50% to 95%; low-level, 5% to 49%; rejection, <5%. MAC resulted in a higher percentage of patients with full donor (>95%) CD3+ T-cell chimerism compared with RIC at day 100 (P < .001), 6 months (P = .006), and 1 year (P = .002) after HCT. This difference was not significant at 2 years after HCT. MAC resulted in a higher percentage of patients with full donor (>95%) B-cell chimerism at day 100 (P = .004) and myeloid chimerism at 6 months (P = .023), compared with RIC. The differences at all other time points were not significant.
Donor chimerism and conditioning intensity
Recipients of MAC vs RIC had significantly different donor T-cell, B-cell, and myeloid engraftment. Specifically, MAC resulted in a higher percentage of patients with full donor (>95%) CD3+ T-cell chimerism than did RIC at day 100 (P < .001), 6 months (P = .006), and 1 year (P = .002) after HCT (Figure 4). This difference was no longer present at 2 years. In addition, MAC resulted in a higher percentage of patients with full donor (>95%) B-cell chimerism at day 100 (P = .004) and myeloid chimerism at 6 months (P = .023), compared with RIC (Figure 4). Differences at all other time points were not significant. We further evaluated whether there was a difference in donor engraftment in recipients of the most commonly used regimens: busulfan-based MAC, busulfan-based RIC, or RIC-other (fludarabine/melphalan ± thiotepa). There was a higher percentage of patients with donor myeloid chimerism ≥50% in recipients of busulfan-based MAC vs RIC-other, as well as busulfan-based RIC vs RIC-other at day 100 (94% vs 60%; P = .009 and 100% vs 60%; P = .009) and 6 months (96% vs 38%; P < .001 and 93% vs 38%; P = .011), respectively. All other comparisons were not significant.
Donor myeloid chimerism and platelet recovery
Following removal of the 8 patients who had a history of a splenectomy before HCT, there were 114 and 102 patients with platelet count data available at day 100 and 1 year after HCT, respectively. Most patients achieved a platelet count of >100 000/μL (n = 79; 69%) or >150 000/μL (n = 63; 55%) by day 100. Thrombocytopenia improved over time with most patients having a platelet count >100 000/μL (n = 91; 89%) or >150 000/μL (n = 81; 79%) at 1 year. However, 11 patients did not achieve a platelet count >100 000/μL at 1 year. Potential causes included low myeloid engraftment/graft rejection (7 patients), GVHD plus infections (2 patients), and an unknown cause (2 patients).
We found that the recovery of platelet counts after HCT was related to the degree of donor myeloid chimerism (Figure 5). Specifically, platelet counts in patients who had low-level (5%-49%) donor myeloid engraftment remained inferior to platelet counts in those who had attained full (>95%) donor myeloid engraftment at 1 year (median, 40 000/μL vs 309 500/μL; P < .0001), 2 years (median, 60 000/μL vs 297 500/μL; P < .0001), and 3 to 5 years (median, 71 000/μL vs 295 000/μL; P = .0002; Figure 5). In addition, platelet counts were lower in patients with low-level compared with high-level (50% to 95%) donor myeloid engraftment at 1 year (median, 40 000/μL vs 205 000/μL; P = .011). All other pairwise comparisons were not significant.
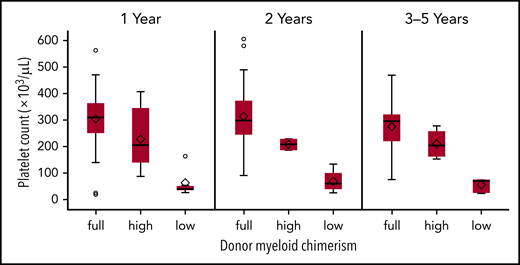
Box plots of platelet count vs donor myeloid chimerism at 1 and 2 years and 3 to 5 years after HCT. Donor myeloid chimerism: full, >95%; high-level, 50% to 95%; low-level, 5% to 49%. Recovery of platelet counts after HCT was related to the degree of donor myeloid chimerism. Specifically, platelet counts for patients who had low-level (5% to 49%) donor myeloid engraftment remained inferior to those who had attained full (>95%) donor myeloid engraftment at 1 year (P < .0001), 2 years (P < .0001), and 3 to 5 years (P = .0002). In addition, platelet counts were lower in patients with low-level (5% to 49%) donor myeloid engraftment compared with those who attained high level (50% to 95%) donor myeloid engraftment at 1 year after HCT (P = .011); however, this was not significant at 2 or 3 to 5 years after HCT. All other pairwise comparisons were not significant.
Box plots of platelet count vs donor myeloid chimerism at 1 and 2 years and 3 to 5 years after HCT. Donor myeloid chimerism: full, >95%; high-level, 50% to 95%; low-level, 5% to 49%. Recovery of platelet counts after HCT was related to the degree of donor myeloid chimerism. Specifically, platelet counts for patients who had low-level (5% to 49%) donor myeloid engraftment remained inferior to those who had attained full (>95%) donor myeloid engraftment at 1 year (P < .0001), 2 years (P < .0001), and 3 to 5 years (P = .0002). In addition, platelet counts were lower in patients with low-level (5% to 49%) donor myeloid engraftment compared with those who attained high level (50% to 95%) donor myeloid engraftment at 1 year after HCT (P = .011); however, this was not significant at 2 or 3 to 5 years after HCT. All other pairwise comparisons were not significant.
Autoimmune disease
Twenty-six patients had a history of autoimmune disease present at the time of conditioning, of whom 25 survived to day 100 after HCT and were therefore evaluable for disease resolution. Of the 25 evaluable patients, 24 resolved the autoimmunity after HCT, and nearly all resolved within the first year after HCT. Specifically, 18 patients had resolution within the first 100 days and an additional 5 had resolution by 1 year. Of the 129 patients, 123 survived to day 100 and were evaluable for the development of de novo autoimmune disease after HCT. Seventeen patients (14%) developed 1 or more de novo autoimmune diseases, all within the first year. Cytopenias were the most common de novo autoimmune diseases after HCT; these included hemolytic anemia (n = 13), thrombocytopenia (n = 6), and neutropenia (n = 3). Most patients (13 of 17) had resolution of de novo autoimmune disease.
We evaluated whether having autoimmune disease before HCT increased the risk for de novo autoimmune disease after HCT and found no association (P = .7792). In addition, we looked at the association of autoimmune disease after HCT and donor type. We found that none of the recipients of HLA-matched sibling grafts developed de novo autoimmune disease, whereas 23% of patients receiving unrelated grafts and 9% with CB grafts developed de novo autoimmune disease. The only significant difference was between HLA-matched sibling grafts and unrelated grafts (P = .0152). All other comparisons were not significant (HLA-matched sibling grafts vs CB grafts [P = .2763] or unrelated grafts vs CB grafts [P = .0958]). There was no difference in the incidence of de novo autoimmune disease based on WAS score or age at HCT (data not shown).
Based on prior reports demonstrating an association between mixed chimerism and autoimmunity, we evaluated whether patients with mixed chimerism were at a higher risk of developing autoimmune diseases after HCT. Specifically, we examined whether mixed-donor chimerism (≤95%) at day 100 or 6 months correlated with an increased risk for the development of de novo autoimmunity at day 100 or later or at 6 months or later, respectively, after HCT. Unlike results published in prior studies,14,16 we did not find an association between mixed donor chimerism and the development of de novo autoimmunity after HCT, except for mixed myeloid donor chimerism at 6 months after HCT (P = .0494; Table 5).
Chimerism and post-HCT autoimmune disease
| Chimerism | No de novo autoimmunity at day 100 or later | De novo autoimmunity at day 100 or later | P |
|---|---|---|---|
| At day 100 | |||
| All chimerism (n = 112)* | .5841 | ||
| Full | 58 (88) | 8 (12) | |
| Mixed | 38 (83) | 8 (17) | |
| T-cell chimerism (n = 84) | .2721 | ||
| Full | 53 (91) | 5 (9) | |
| Mixed | 21 (81) | 5 (19) | |
| B-cell chimerism (n = 59) | .1299 | ||
| Full | 44 (88) | 6 (12) | |
| Mixed | 6 (67) | 3 (33) | |
| Myeloid chimerism (n = 83) | .6802 | ||
| Full | 57 (90) | 6 (10) | |
| Mixed | 17 (85) | 3 (15) |
| Chimerism | No de novo autoimmunity at day 100 or later | De novo autoimmunity at day 100 or later | P |
|---|---|---|---|
| At day 100 | |||
| All chimerism (n = 112)* | .5841 | ||
| Full | 58 (88) | 8 (12) | |
| Mixed | 38 (83) | 8 (17) | |
| T-cell chimerism (n = 84) | .2721 | ||
| Full | 53 (91) | 5 (9) | |
| Mixed | 21 (81) | 5 (19) | |
| B-cell chimerism (n = 59) | .1299 | ||
| Full | 44 (88) | 6 (12) | |
| Mixed | 6 (67) | 3 (33) | |
| Myeloid chimerism (n = 83) | .6802 | ||
| Full | 57 (90) | 6 (10) | |
| Mixed | 17 (85) | 3 (15) |
| Chimerism | No de novo autoimmunity at 6 mo or later | De novo autoimmunity at 6 mo or later | P |
|---|---|---|---|
| At 6 mo | |||
| All chimerism (n = 82)* | .3938 | ||
| Full | 45 (96) | 2 (4) | |
| Mixed | 31 (89) | 4 (11) | |
| T-cell chimerism (n = 65) | .5995 | ||
| Full | 45 (94) | 3 (6) | |
| Mixed | 15 (88) | 2 (12) | |
| B-cell chimerism (n = 48) | .2865 | ||
| Full | 41 (95) | 2 (5) | |
| Mixed | 4 (80) | 1 (20) | |
| Myeloid chimerism (n = 62) | .0494 | ||
| Full | 45 (98) | 1 (2) | |
| Mixed | 13 (81) | 3 (19) |
| Chimerism | No de novo autoimmunity at 6 mo or later | De novo autoimmunity at 6 mo or later | P |
|---|---|---|---|
| At 6 mo | |||
| All chimerism (n = 82)* | .3938 | ||
| Full | 45 (96) | 2 (4) | |
| Mixed | 31 (89) | 4 (11) | |
| T-cell chimerism (n = 65) | .5995 | ||
| Full | 45 (94) | 3 (6) | |
| Mixed | 15 (88) | 2 (12) | |
| B-cell chimerism (n = 48) | .2865 | ||
| Full | 41 (95) | 2 (5) | |
| Mixed | 4 (80) | 1 (20) | |
| Myeloid chimerism (n = 62) | .0494 | ||
| Full | 45 (98) | 1 (2) | |
| Mixed | 13 (81) | 3 (19) |
Data are expressed as the number of recipients (percentage of total in chimerism subgroup).
All chimerism was defined as any chimerism lineage measured (T-cell, B-cell, myeloid, whole-blood, or peripheral blood mononuclear cell). If a patient had mixed chimerism (≤95%) in any lineage, it was coded as mixed. The 6-month analysis of autoimmune disease included only patients who were alive and did not have de novo autoimmune disease prior to 6 months after HCT.
Discussion
This is the largest multicenter, retrospective study of HCT outcomes of patients with WAS who underwent HCT in North America. OS after HCT was excellent regardless of HCT year, WAS score, WASP expression, conditioning regimen intensity, donor type, and hematopoietic cell source. Supporting what has been previously reported,13,16 our study demonstrated that age remained an important factor for OS. Specifically, 5-year OS was superior in patients who were <5 years vs ≥5 years of age at the time of HCT (94% vs 66%, respectively; overall P = .0008). Our cohort had a high number of infants who underwent HCT (n = 53; <1 year of age) with excellent 5-year OS (94%). The high proportion of patients transplanted in infancy was not related to a more severe clinical course before HCT. Rather, this could reflect a trend in earlier and faster definitive diagnosis, broader donor availability (with the use of CB grafts), an awareness that the WAS score in patients <2 years of age is not predictive of overall disease severity, and prior reports demonstrating superior OS in patients who underwent HCT before 5 years of age13 and, more recently, before 2 years of age.16
There are limited published outcome data after CB HCT for patients with WAS. Shekhovtsova et al27 reported on 90 patients with WAS who underwent CB HCT after primarily myeloablative conditioning. At 5 years, the OS was 75% with superior OS in patients ≤2 years of age at the time of HCT (5-year OS, 83%). Moratto et al16 reported HCT outcomes of 194 patients with WAS, of whom 24 (12%) received CB grafts, with inferior survival observed in recipients of CB vs HLA-matched sibling grafts. In contrast, in our study of 129 patients, of whom 39 (30%) received CB grafts, we found no significant difference in OS based on donor type (HLA-matched sibling, unrelated donor, or unrelated CB) and with excellent 5-year OS of 90% after CB HCT. This may in part be related to the high number of well-matched CB grafts in our study (6 of 6 [n = 13; 33%] and 5 of 6 [n = 20; 51%]). In addition, our study included only patients who underwent HCT during a more recent period (2005–2015). Survival after CB HCT has improved over time because of better CB selection based on degree of HLA-match, higher CB cell dose, and improved supportive care after HCT. These factors likely contributed to the excellent OS we observed after CB HCT.
In previous retrospective studies, more patients received TCD-mismatched related grafts than in our cohort and had less favorable outcomes. In our cohort, only 1 patient underwent HCT using a TCD-mismatched related graft, which may reflect the poor historic outcomes. More recently, HCT using post-HCT cyclophosphamide or TCRαβ/CD19-depleted haploidentical grafts have yielded favorable results in patients with nonmalignant diseases, including WAS.28-36 Autologous gene therapy is a promising investigational approach that has the advantage of avoiding GVHD and rejection. Although the initial trial using a gammaretroviral vector led to a high rate of insertional oncogenesis,37,38 trials using a self-inactivating lentiviral vector in which the WAS gene is under the control of a 1.6-kb human WAS promoter have shown encouraging efficacy with no evidence of insertional oncogenesis to date.39,40 However, platelet reconstitution was achieved less consistently, and comparison in the future with allogeneic HCT recipients would be valuable.
Similar to other reports,16,41 we found a strong association between donor myeloid engraftment and platelet recovery. Specifically, higher platelet counts were observed among recipients who achieved full (>95%) vs those with low-level (5%-49%) donor myeloid engraftment at 1 year, 2 years, and 3 to 5 years after HCT. Donor myeloid chimerism of <50% was associated with thrombocytopenia, with median platelet counts of 40 000/μL (1-year), 60 000/μL (year 2), and 71 000/μL (3-5 years).
Importantly, because our cohort included a large number of patients who received RIC, we were able to compare the impact on engraftment of MAC vs RIC and of different RIC regimens. We found that MAC (almost exclusively busulfan and cyclophosphamide) was associated with improved T-cell engraftment within the first year compared with RIC. In addition, MAC resulted in a higher percentage of patients with full donor B-cell chimerism at day 100 and myeloid chimerism at 6 months compared with RIC; however, differences at all other time points were not significant. This may in part have been due to the low number of patients with chimerism data available for analysis at later time points after HCT, which highlights the importance of prospective studies that rigorously capture long-term engraftment data in a larger number of patients after HCT.
We also looked at whether there was a difference in MAC or RIC busulfan-based regimens vs RIC-other (fludarabine/melphalan-containing regimens). Busulfan-based MAC and busulfan-based RIC resulted in a higher percentage of patients with donor myeloid engraftment ≥50% early after HCT, compared with RIC-other. Thus, our data suggest that historic MAC regimens such as busulfan combined with cyclophosphamide may not be the only means of achieving engraftment and disease correction. The difference we saw between RIC regimens, with fludarabine/melphalan-containing regimens resulting in inferior myeloid engraftment early after HCT, points to the need to develop regimens that achieve robust engraftment similar to busulfan-based RIC with fewer short- and long-term toxicities. Our definitions of MAC and RIC notwithstanding, there is likely to be a continuum of intensity, with some RIC regimens being more accurately categorized as myeloablative, but with reduced toxicity (for example, treosulfan-based regimens), which were not represented in our study. Therefore, prospective studies are needed using reduced-intensity or reduced-toxicity conditioning to better evaluate long-term engraftment and disease response in patients with WAS.
Several retrospective studies have reported an association between mixed-donor chimerism and autoimmunity after HCT.14,16 Our study did not find such an association, except at 6 months after HCT in patients with mixed-donor myeloid chimerism. This may have been due to the relatively low incidence of autoimmunity after HCT in our cohort; therefore, our results should be interpreted with caution. However, it should be noted that Shin et al17 and Iguchi et al42 also did not find an association between mixed chimerism and autoimmune disease after HCT. Therefore, prospective studies are needed to better address this question. One observation that is important to highlight is that, in those patients with a history of autoimmune disease at time of conditioning, nearly all had resolution of the autoimmune disease within 1 year after HCT. In addition, for reasons that are not completely understood, we found an association between donor type and the development of autoimmune disease after HCT. Specifically, we found a higher incidence of de novo autoimmune disease among recipients of unrelated grafts vs HLA-matched sibling grafts (P = .0152). All other comparisons among donor types and de novo autoimmune disease were not significant.
In conclusion, our retrospective analysis demonstrated excellent OS after HCT, independent of donor type, hematopoietic cell source, or WAS score. HCT at a younger age continues to be associated with superior outcomes, supporting the recommendation for HCT early in the course of the disease before comorbidities develop. Importantly, HCT should not be delayed if a suitable related, unrelated, or CB donor can be identified. Donor myeloid engraftment ≥50% was associated with higher platelet counts after HCT. Although OS in those undergoing HCT at <1 year of age was excellent, the potential disadvantages of proceeding in the first year of life should be examined. Future studies should evaluate the incidence of long-term complications after HCT in patients with WAS, including infertility, secondary malignancies, endocrine effects, and organ dysfunction, and examine whether conditioning regimen and/or early age at HCT contribute to these complications. Equally important, studies are needed to better evaluate quality of life (QoL) after HCT in patients with WAS. Shah et al43 recently reported on improved QoL for patients after HCT according to their parents; however, patients reported no difference in QoL among those who underwent HCT vs those who did not. Shah et al were among the first to look at QoL after HCT for WAS, and they highlighted the importance of future prospective studies to better evaluate QoL after HCT in WAS survivors.
Please contact Lauri M. Burroughs (lburroug@fredhutch.org) regarding protocol details.
For original data, please e-mail the corresponding author.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Catherine Chang, Tara Bani, and Elizabeth Dunn for project management and assistance; all of the study coordinators for collection of clinical data from PIDTC sites and the clinical care teams who provided care for patients; Sumathi Iyengar, Executive Director of the Wiskott-Aldrich Foundation, and all of the patients and families who have made this work possible; and Helen Crawford for her assistance with manuscript and figure preparation.
This work was supported by the Division of Allergy, Immunology and Transplantation, National Institute of Allergy and Infectious Diseases (NIAID); the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH); Public Health Service, NIH, NIAID grant U54AI082973 (principal investigator [PI], M.J.C.; PIs from September 2019 forward, J.M.P. and D.B.K.); NIH, National Institute of Neurological Disorders and Stroke grant U54NS064808 and National Center for Advancing Translational Sciences grant U01TR001263 (PI, J. P. Krischer); NIH, NIAID grant R13AI094943 (PIs, M.J.C. and March 2018 forward, J.M.P.). The PIDTC is a part of the Rare Diseases Clinical Research Network (RDCRN) of ORDR, NCATS. The collaborative work of the PIDTC with the Pediatric Blood and Marrow Transplant Consortium (PBMTC) is supported by the U54 grants listed here, along with support of the PBMTC Operations Center by the St. Baldrick’s Foundation and NIH, National Heart, Lung and Blood Institute (NHLBI) grant/cooperative agreement U10HL069254 (PI, M.A.P.). Collaborative work of the PIDTC with the Center for International Blood and Marrow Transplant Research (CIBMTR) is supported by NIH, National Cancer Institute grant/cooperative agreement U24CA076518 (PI, M. M. Horowitz), and NHLBI, and NIAID; and NIH, NHLBI grant/cooperative agreement U01HL069294; by contracts HHSH250201200016C and HHSH234200637015C with the Health Resources and Services Administration (HRSA/DHHS); and by grants N00014-13-1-0039 and N00014-14-1-0028 from the Office of Naval Research. In addition, the Dejoria Wiskott-Aldrich Research Fund and the Jeffrey Modell Foundation helped fund the mutation analyses and WASP expression studies for many of the patients included in this cohort. L.D.N. is supported by the Division of Intramural Research, NIH, NIAID grant 1 ZIA AI001222-02 (PI, L.D.N.). S.S. is supported by the Children’s Discovery Institute, St. Louis Children’s Hospital.
Authorship
Contribution: L.M.B., L.M.G., L.D.N., D.J.R., and M.J.C. designed the study; L.M.B., A.P., R.B., and X.L. wrote the first draft of the manuscript; R.B. and X.L. performed the statistical analyses; L.M.B., A.P., L.M.G., H.D.O., S.E.P., S.C., A.R.S., S.P., K.G.W., M.D.K., J.R., E.H., K.E.S., D.B.K., J.M.P., L.D.N., S.-Y.P., and M.J.C. reviewed each submitted case to assess protocol eligibility and/or served on the Protocol 6904 WAS committee, cleaned the data, and contributed to the data analysis; M.J.C., J.M.P, D.B.K., S.-Y.P., E.H., M.A.P., T.T., L.D.N., and L.M.G. gave overall direction as members of the PIDTC Steering Committee; L.M.B., A.P., R.B., X.L., L.M.G., H.D.O., J.J.B., S.E., C.C.D., S. Chaudhury, S.E.P., R.Q., F.D.G., T.C.Q., S. Chandrakasan, A.R.S., S.P., B.J.D.S., M.S.T., R.P., S.S., L.R.F., C.M., D.C., E.S., H.K.M., N.K., J.L.B., H.C., D.C.S., K.C., R.A.-A., A.J.S., K.G.W., T.B.M., A.J., K.B.D., A.P.G., G.D.E.C., M.D.K., J.R., T.T., M.A.P., E.H., K.E.S, B.R.L., D.B.K., J.M.P., L.D.N., S.-Y.P., D.J.R., and M.J.C. identified patients for the study and/or edited the manuscript; L.M.G., an employee of the Division of Allergy, Immunology, and Transplantation, NIAID, NIH, the funding source, also participated as a PIDTC study team member in the interpretation of data, writing of the manuscript, and decision to submit the manuscript for publication; and all authors edited the manuscript and contributed to the subsequent versions.
Conflict-of-interest disclosure: L.M.B. received support for the conduct of a clinical trial through the Fred Hutchinson Cancer Research Center by Medac GmbH, including supply of the study drug Treosulfan. S. Chaudhury is on the Mesoblast Advisory Panel. S.E.P. receives support for the conduct of sponsored trials through Memorial Sloan Kettering from Atara Biotherapeutics and Mesoblast Advisory Panel Mesoblast. R.P. is on the advisory board for Orchard Therapeutics. M.D.K. is on the advisory board for Gilead Sciences. The remaining authors declare no competing financial interests.
The content and opinions expressed are solely the responsibility of the authors and do not represent the official policy or position of the NIAID, ORDR, NCATS, NIH, HRSA, or any other agency of the US Government.
Correspondence: Lauri M. Burroughs, Fred Hutchinson Cancer Research Center, 1100 Fairview Ave North, Suite D5-289, PO Box 19024, Seattle, WA 98109-1024; e-mail: lburroug@fredhutch.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal