


Abstract
Langerhans cell histiocytosis (LCH) is caused by clonal expansion of myeloid precursors that differentiate into CD1a+/CD207+ cells in lesions that leads to a spectrum of organ involvement and dysfunction. The pathogenic cells are defined by constitutive activation of the MAPK signaling pathway. Treatment of LCH is risk-adapted: patients with single lesions may respond well to local treatment, whereas patients with multisystem disease require systemic therapy. Although survival rates for patients without organ dysfunction is excellent, mortality rates for patients with organ dysfunction may reach 20%. Despite progress made in the treatment of LCH, disease reactivation rates remain above 30%, and standard second-line treatment is yet to be established. Treatment failure is associated with increased risks for death and long-term morbidity, including LCH-associated neurodegeneration. Early case series report promising clinical responses in patients with relapsed and refractory LCH treated with BRAF or MEK inhibitors, although potential for this strategy to achieve cure remains uncertain.
Introduction
Langerhans cell histiocytosis (LCH) is a disease characterized by clonal expansion of myeloid precursors that differentiate into CD1a+/CD207+ in lesions. It presents at all ages with various degrees of systemic involvement, and although cure rates are high, severe long-term neurological or endocrine complications may affect quality of life. Our understanding of the pathogenesis of the disease has evolved from a reactive clonal proliferation of Langerhans cells (LCs) to an inflammatory myeloid neoplasia; this evolution from a disorder of immune dysregulation to a bona fide neoplastic disorder has reclassified the disease and opened the door for the development of targeted therapies.
Epidemiology
The reported incidence of LCH ranges from 2.6 to 8.9 cases per million children younger than 15 years per year, with a median age at diagnosis of 3 years.1-4 The exact incidence of LCH in adults is much less defined: the only available data are for disseminated disease, with 0.07 cases per million per year.5,6
The causes and risk factors for developing LCH are unclear.7 Population-based studies have shown differences in the incidence of multisystem LCH by race and ethnicity; in children, higher incidence has been reported for Hispanics, and lower for blacks.8 The lower incidence in the black population has also been reported in adults.6 In a population-based, case-control study, Hispanic mothers were more likely to have children who developed LCH compared with non-Hispanic whites; this risk increased when both parents were Hispanic. Non-Hispanic black mothers were less likely to give birth to offspring who developed LCH compared with non-Hispanic whites.9 The association with Hispanic ancestry has been further documented in a genome-wide association study, which identified a novel risk variant within SMAD6.10 The observation of increased incidence in monozygotic twins of affected patients has also suggested the presence of a germline predisposition, at least for a small proportion of cases.11
The association between LCH and other malignancies has been described, with frequencies varying from 2.6% in children12 to 32% in adults.13 Different solid malignancies have been associated with LCH; lung carcinoma has been consistently reported in adult series,13,14 and thyroid carcinoma has been noted to occur in conjunction with thyroid infiltration by LCH in both adults and children.15,16 Hodgkin and non-Hodgkin lymphomas have been described in association with LCH, often occurring concurrently in the same nodes.14,17 The most common hematologic malignancy reported is acute myeloid leukemia, often occurring years after LCH.12-14,18 In contrast, LCH in association with acute lymphoblastic leukemia (ALL) commonly occurs during treatment.18-20 Reports have shown that coincident LCH and ALL share the same oncogenic mutations or have an identical T-cell receptor or immunoglobulin rearrangement, suggesting the presence of a clonal relationship between LCH and ALL.19,20
Pathology
More than 100 different histiocytic disorders have been described, and a classification system consisting of 5 groups of diseases has been proposed: LCH-related, cutaneous and mucocutaneous non-LCH histiocytoses, Rosai-Dorfman disease, malignant histiocytoses, and hemophagocytic lymphohistiocytosis and macrophage activation syndrome (Table 1).21
Classification of histiocytoses
| Histiocytosis group | Diseases |
|---|---|
| L Group | LCH |
| Indeterminate-cell histiocytosis (ICH) | |
| Erdheim-Chester Disease (ECD) | |
| Mixed LCH/ECD | |
| C Group | Cutaneous non-LCH |
| Xanthomatous granuloma (XG) family: JXG, AXG, SRH, BCH, GEH, PNH | |
| Non-XG family: cutaneous RDD, NXG, other | |
| Cutaneous non-LCH with a major systemic component | |
| XG family: XD | |
| Non-XG family: MRH | |
| R Group | Familial RDD |
| Sporadic RDD | |
| Classical RDD | |
| Extranodal RDD | |
| RDD with neoplasia or immune disease | |
| Unclassified | |
| M Group | Primary Malignant Histiocytoses |
| Secondary Malignant Histiocytoses | |
| H Group | Primary HLH: Monogenic inherited conditions leading to HLH |
| Secondary HLH (non-Mendelian HLH) | |
| HLH of unknown/uncertain origin |
| Histiocytosis group | Diseases |
|---|---|
| L Group | LCH |
| Indeterminate-cell histiocytosis (ICH) | |
| Erdheim-Chester Disease (ECD) | |
| Mixed LCH/ECD | |
| C Group | Cutaneous non-LCH |
| Xanthomatous granuloma (XG) family: JXG, AXG, SRH, BCH, GEH, PNH | |
| Non-XG family: cutaneous RDD, NXG, other | |
| Cutaneous non-LCH with a major systemic component | |
| XG family: XD | |
| Non-XG family: MRH | |
| R Group | Familial RDD |
| Sporadic RDD | |
| Classical RDD | |
| Extranodal RDD | |
| RDD with neoplasia or immune disease | |
| Unclassified | |
| M Group | Primary Malignant Histiocytoses |
| Secondary Malignant Histiocytoses | |
| H Group | Primary HLH: Monogenic inherited conditions leading to HLH |
| Secondary HLH (non-Mendelian HLH) | |
| HLH of unknown/uncertain origin |
Adapted from Emile et al.21
AXG, adult xanthogranuloma; BCH, benign cephalic histiocytosis; GEH, generalized eruptive histiocytosis; JXG, juvenile xanthogranuloma; MRH, multicentric reticulohistiocytosis; NXG, necrobiotic xanthogranuloma; PNH, progressive nodular histiocytosis; RDD, Rosai-Dorfman Disease; SRH, solitary reticulohistiocytoma; XD, xanthoma disseminatum.
Diagnosis of LCH requires a clonal neoplastic proliferation with expression of CD1a, CD207 (Langerin), and S100 (Figure 1). The cells are generally large, round to oval in shape, with a coffee-bean nuclear grove, and without the branching that characterizes inflammatory CD1a+ dendritic cells. On electron microscopy, pentalaminar cytoplasmic rod-shaped inclusions (Birbeck granules) can be identified, although electron microscopy is no longer required for diagnosis in the presence of CD207+ staining. Because LCH cells activate and recruit other immunologic cells, microscopic examination shows an inflammatory pattern consisting of eosinophils, neutrophils, lymphocytes, and macrophages in addition to the LCs; this appearance is what is described as eosinophilic granuloma.22
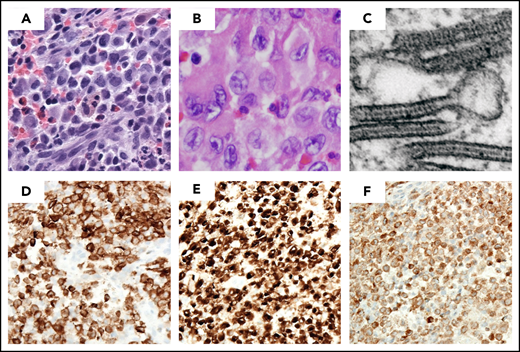
LCH lesion histology. Images demonstrate typical histology of LCH lesion obtained from a bone biopsy with pathologic histiocytes and inflammatory infiltrate. (A-B) Hematoxylin and eosin stain demonstrates histiocytes with pale cytoplasm and reniform nuclei. (C) Birbeck granules on electron microscopy; immunohistochemistry strongly positive for (D) langerin/CD207, (E) CD1a, and (F) S100a. (Courtesy of M. J. Hicks)
LCH lesion histology. Images demonstrate typical histology of LCH lesion obtained from a bone biopsy with pathologic histiocytes and inflammatory infiltrate. (A-B) Hematoxylin and eosin stain demonstrates histiocytes with pale cytoplasm and reniform nuclei. (C) Birbeck granules on electron microscopy; immunohistochemistry strongly positive for (D) langerin/CD207, (E) CD1a, and (F) S100a. (Courtesy of M. J. Hicks)
Clinical presentation
LCH presents in a continuum of systemic involvement, ranging from a solitary eosinophilic granuloma to widespread disseminated disease with organ dysfunction.23 The current classification system is based on the site of lesions, number of involved sites (single or multisystem/local or multifocal), and whether the disease is involving risk (of mortality) organs (hematopoietic system, liver, or spleen; Tables 2 and 3). In a large cohort review, single-system and multisystem disease accounted for approximately half of the patients each. Among the patients with multisystem disease, approximately 15% of them had involvement of a risk organ.24 The skeleton is the most commonly affected system, as bone lesions are present in approximately 80% of patients with LCH,25 and in half of them, lesions are single.24 The most common site of bone involvement is the skull, followed by spine, limbs, and pelvis.26
Clinical Classification of LCH
| Clinical group | Description |
|---|---|
| Multisystem | Two or more systems involved |
| With risk organ involvement | Involvement of liver, spleen or bone marrow |
| Without risk organ involvement | Without involvement of liver, spleen or bone marrow |
| Single-system | Only 1 system involved |
| Single site | Skin, bone, lymph node, other (thyroid, thymus) |
| Multiple sites | Multifocal bone disease |
| Special site | Skull-base lesion with intracranial extension or vertebral lesion with intraspinal soft tissue extension |
| Pulmonary LCH | Isolated lung disease |
| CNS LCH | Tumorous lesions |
| Neurodegenerative disease | |
| LACI | |
| LACS |
| Clinical group | Description |
|---|---|
| Multisystem | Two or more systems involved |
| With risk organ involvement | Involvement of liver, spleen or bone marrow |
| Without risk organ involvement | Without involvement of liver, spleen or bone marrow |
| Single-system | Only 1 system involved |
| Single site | Skin, bone, lymph node, other (thyroid, thymus) |
| Multiple sites | Multifocal bone disease |
| Special site | Skull-base lesion with intracranial extension or vertebral lesion with intraspinal soft tissue extension |
| Pulmonary LCH | Isolated lung disease |
| CNS LCH | Tumorous lesions |
| Neurodegenerative disease | |
| LACI | |
| LACS |
Definition of risk organ involvement in LCH-IV
| Hematopoietic involvement (with or without bone marrow involvement*), at least 2 of the following: |
| Anemia: hemoglobin <100 g/L (<10 g/dl), infants <90 g/L (<9.0 g/dL), not a result of other causes (eg, iron deficiency) |
| Leukocytopenia: leukocytes <4.0 × 109/L (4000/μL) |
| Thrombocytopenia: platelets <100 × 109/L (100 000/μL) |
| Spleen involvement enlargement: |
| >2 cm below costal margin in the midclavicular line† |
| Liver involvement, one or more of the following: |
| Enlargement >3 cm below costal margin in the midclavicular line† |
| Dysfunction (ie, hypoproteinemia <55 g/L, hypoalbuminemia <25 g/L, not as a result of other causes) |
| Histopathological findings of active disease |
| Hematopoietic involvement (with or without bone marrow involvement*), at least 2 of the following: |
| Anemia: hemoglobin <100 g/L (<10 g/dl), infants <90 g/L (<9.0 g/dL), not a result of other causes (eg, iron deficiency) |
| Leukocytopenia: leukocytes <4.0 × 109/L (4000/μL) |
| Thrombocytopenia: platelets <100 × 109/L (100 000/μL) |
| Spleen involvement enlargement: |
| >2 cm below costal margin in the midclavicular line† |
| Liver involvement, one or more of the following: |
| Enlargement >3 cm below costal margin in the midclavicular line† |
| Dysfunction (ie, hypoproteinemia <55 g/L, hypoalbuminemia <25 g/L, not as a result of other causes) |
| Histopathological findings of active disease |
Bone marrow involvement is defined as presence of CD1a positive cells on marrow slides.
Enlargement in centimeters below the costal margin as assessed by physical examination.
Most organs can be affected by LCH, and therefore a comprehensive evaluation is indicated.27 Symptoms and physical and laboratory examination should guide the extent of diagnostic studies; focus should be on assessing the number of systems and sites involved, and on the involvement of risk organs. Bone imaging studies reveal a lytic lesion without marginal sclerosis, with or without periosteal reaction. (Figure 2) Radio-isotope imaging is recommended to assess the number of bone lesions; fluorodeoxyglucose–positron emission tomography scans can be useful in defining the extent of the disease and the response to therapy.28,29 (Figure 2) The skull, including the skull base, is very commonly involved; typical locations include the bones of the orbit or the temporal bone (typically the mastoid). Involvement of the vertebral bodies is also common, and the presence of a vertebra plana is frequent. Pain and tumor formation in a localized area of bone is a very common presentation of LCH.
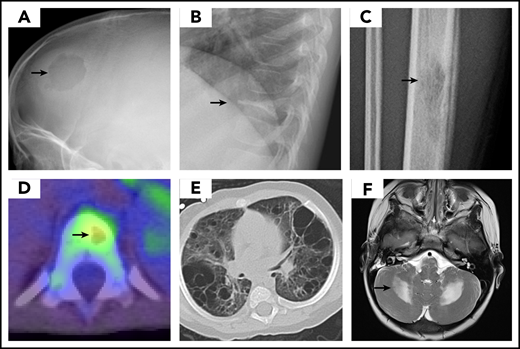
LCH imaging. Images demonstrate typical presentation of LCH lesions including (A) single skull lesion on X-ray, (B) vertebra plana on X-ray, (C) femur lesion on X-ray, (D) vertebral lesion on positron emission tomography/computed tomography scan, and (E) cystic lung disease on computed tomography scan. (F) Brain magnetic resonance imaging demonstrates T2 hyperintensity in cerebellum in LCH-associated neurodegeneration. (Courtesy of P. Campbell)
LCH imaging. Images demonstrate typical presentation of LCH lesions including (A) single skull lesion on X-ray, (B) vertebra plana on X-ray, (C) femur lesion on X-ray, (D) vertebral lesion on positron emission tomography/computed tomography scan, and (E) cystic lung disease on computed tomography scan. (F) Brain magnetic resonance imaging demonstrates T2 hyperintensity in cerebellum in LCH-associated neurodegeneration. (Courtesy of P. Campbell)
Skin involvement is also common, particularly in infants, where it presents as seborrheic eczema, and in adults, where it may present as refractory eczema in intertriginous and genital areas (Figure 3). Isolated skin involvement usually carries a good prognosis, with an approximately 60% chance of regression with topical treatments.30-34 The lesions of congenital self-healing LCH are often present at or shortly after birth; can appear as eroded or ulcerated papules, pustules, or vesicles with hemorrhagic crusting; and may masquerade as diffuse neonatal hemangiomatosis33 or blueberry muffin rash.34 Close monitoring is required in infants, as reactivation or progression to multisystem involvement has been observed in up to 40% of cases.35,36 In adults, cutaneous involvement commonly presents as papules and intertrigo, with significant scaling and crusting, most commonly in the scalp, although mucosal involvement of the genitalia or oral cavity is also common.37-39
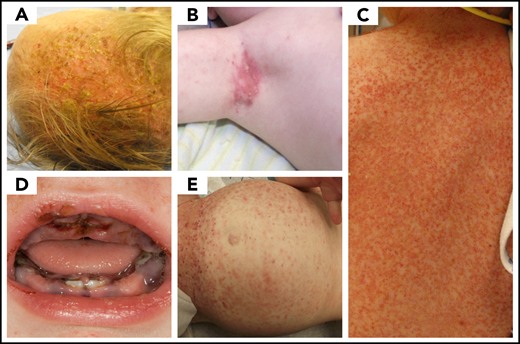
LCH skin disease. A range of LCH skin disease is presented. Rashes and skin lesions are pleotropic in LCH and can mimic other more common pediatric rashes associated with (A) cradle cap, (B) eczema, (C) scarlet fever or scalded skin syndrome, (D) herpes gingivostomatitis, and (E) immune thrombocytopenic purpura. (Courtesy of P. Campbell)
LCH skin disease. A range of LCH skin disease is presented. Rashes and skin lesions are pleotropic in LCH and can mimic other more common pediatric rashes associated with (A) cradle cap, (B) eczema, (C) scarlet fever or scalded skin syndrome, (D) herpes gingivostomatitis, and (E) immune thrombocytopenic purpura. (Courtesy of P. Campbell)
In children, lung involvement usually occurs in the context of multisystem disease, where it has been reported to occur in up to 35% of patients.40 Radiographic findings are typical for the presence of a reticulonodular pattern with bullae formation (Figure 2). In the absence of other risk organ involvement, pulmonary disease is not a predictor of adverse outcome.41-43 Isolated pulmonary involvement is a rare presentation that is almost exclusive of adults with a smoking habit.44
The presence of hematopoietic dysfunction in the form of cytopenias is a poor prognostic sign. It occurs in the context of multisystem involvement, usually in very young children. Its pathophysiology is multifactorial, including direct involvement of the bone marrow as well as peripheral destruction resulting from hypersplenism from LC infiltrates in the spleen.45
Liver involvement also carries a very poor prognosis. Patients present with hypoalbuminemia, hepatomegaly, or conjugated hyperbilirubinemia. A well-described but rare complication in young children and in adults is the development of sclerosing cholangitis and hepatic fibrosis, which commonly evolve to end-stage liver failure.46-48
LCH in Adults
Adult LCH usually presents after the fourth decade, and approximately two-thirds of patients have multisystem involvement at diagnosis.49 Its association with other neoplastic diseases is common, especially other myeloproliferative neoplasms.13 In a significant proportion of patients, LCH and Erdheim-Chester lesions may coexist.50 In general, the clinical presentation and organs involved are similar to pediatric patients, with the more frequent involvement of the genitalia, particularly in females.37,49 The rarity of LCH in adults, combined with the nonspecific and varied clinical presentations, typically result in missed and delayed diagnosis.51,52
Pulmonary LCH
Isolated pulmonary LCH (PLCH) is primarily a disease of young adult smokers, with more than 90% of patients endorsing a smoking history.53-55 PLCH presents with respiratory symptoms, mainly cough and dyspnea on exertion, in approximately two-thirds of the cases. Less frequently, patients may present with spontaneous pneumothorax or with asymptomatic lesions on routine chest X-ray.56 Extrapulmonary organ involvement occurs in 10% to 15% of patients.54 High-resolution CT shows a pattern of bilateral reticulonodular and cystic changes, with apical and midlung predominance, sparing the bases and costophrenic angles.56 Transbronchial lung biopsies may be diagnostic of PLCH in expert centers; however, because of the focal nature of the disease, the diagnostic yield varies between 15% and 40%, and a thoracoscopic lung biopsy is usually recommended.56
Pathology of PLCH shows nodular lesions with the typical histology. In late disease, nodules are replaced by advanced bullous and cystic lesions, often in association with hyperinflation and honeycombing.57 BRAFV600E and MAP2K1 mutations have been reported at similar frequency as in extrapulmonary LCH, although lower mutation rates are identified in the more fibrotic lesions.58,59
LCH of the central nervous system
Central nervous system (CNS) involvement in LCH (LCH-CNS) represents a spectrum of diseases ranging from active infiltration by LCH to long-term effects. Its prevalence has been noted to range from 3.4% to 57%.60 LCH-CNS can be divided in focal mass lesions and lesions associated with progressive neurodegeneration.60,61
Mass lesions tylically present in meninges, choroid plexus, and brain parenchyma. Characteristic neuroimaging findings include hypothalamic-pituitary involvement, often with diabetes insipidus, infundibular thickening, and absent bright spot in posterior pituitary; enlargement and enhancement of the pineal gland; thickening and enhancement of choroid plexus; or intraparenchymal masses.61 Among patients with anterior pituitary dysfunction, the most common deficiency is in antidiuretic hormone, followed by growth hormone (which occurs in up to 50% of patients with diabetes insipidus), gonadotropin, and thyrotropin.62,63 Anterior pituitary dysfunction is more common in childhood-onset patients and in those with multisystem disease.12,62 Diabetes insipidus, the hallmark of this dysfunction, has been reported to occur in up to 24% of patients with LCH, but in half of patients with multisystem disease12,64 ; in one third of cases, the diabetes insipidus precedes or is concurrent with the diagnosis of LCH, and in the remaining two-thirds of the cases, it is diagnosed later.12,40,65 With the use of more comprehensive risk-adapted management of LCH, the incidence of endocrinopathies has decreased to 10% to 15% in recent large cohort studies.24,66
Neurodegenerative LCH (LCH-ND) is characterized by progressive radiologic and clinical abnormalities. As recently reviewed by Yeh et al,60 2 separate clinical forms are identified: LCH-associated abnormal CNS imaging (LACI), which includes asymptomatic patients with radiologic findings, and LCH-associated abnormal CNS symptoms (LACS), which describes patients with abnormal cognitive and psychological findings. LACI and LACS are associated with increased T2-weighted MRI signal in the dentate nucleus of the cerebellum, basal ganglia, and pons (Figure 2; Table 4).
Clinical, histological, and imaging characteristics of LCH of the central nervous system
| Lesion type and site | Pathology | MRI characteristics |
|---|---|---|
| Tumorous lesions | ||
| Cerebral white and gray matter | Typical LCH morphology with CD1a/CD207 + histiocytes | Nodular or space-occupying lesions; T2 hyperintensity and T1 iso- or hypointensity; variably contrast enhancing; can present mass effect |
| LACI | ||
| Dentate nuclei of the cerebellum | Loss of Purkinje cells with gliosis in the cerebellar cortex | Bilateral and symmetrical slight T1-w hyperintensity, followed by development of T1-w hypointensity and/or T2-w hyperintensity |
| Infratentorial white matter (cerebellum, brainstem) | Neuroaxonal loss with secondary demyelination; pronounced inflammatory process dominated by CD8+ T-lymphocytes and microglial activation; BRAFV600E+ perivascular myeloid cells and increased frequency of BRAFV600E+ peripheral blood mononuclear cells (for patients with BRAFV600E+ systemic LCH) | Bilateral and symmetrical abnormalities (T2-w hyperintensity, T1-w isointensity or hypointensity) |
| Basal ganglia | — | Bilateral and symmetrical leukoencephalopathy-like abnormalities, or confluent lesions in a vascular pattern, with T2 hyperintensity and T1 hypointensity |
| Supratentorial white matter | Reactive gliosis and microglial activation decreased BRAFV600E+ cells compared with cerebellum/brainstem | Bilateral and symmetrical leukoencephalopathy-like abnormalities, or confluent lesions in a vascular pattern, with T2 hyperintensity and T1 hypointensity |
| Prominent, dilated perivascular spaces | ||
| Cerebral white matter | — | Bilateral and symmetrical punctate lesions in a vascular pattern. T2-w hyperintensity, and T1 iso- or hypointensity; variable contrast enhancement and mass effect |
| Lesion type and site | Pathology | MRI characteristics |
|---|---|---|
| Tumorous lesions | ||
| Cerebral white and gray matter | Typical LCH morphology with CD1a/CD207 + histiocytes | Nodular or space-occupying lesions; T2 hyperintensity and T1 iso- or hypointensity; variably contrast enhancing; can present mass effect |
| LACI | ||
| Dentate nuclei of the cerebellum | Loss of Purkinje cells with gliosis in the cerebellar cortex | Bilateral and symmetrical slight T1-w hyperintensity, followed by development of T1-w hypointensity and/or T2-w hyperintensity |
| Infratentorial white matter (cerebellum, brainstem) | Neuroaxonal loss with secondary demyelination; pronounced inflammatory process dominated by CD8+ T-lymphocytes and microglial activation; BRAFV600E+ perivascular myeloid cells and increased frequency of BRAFV600E+ peripheral blood mononuclear cells (for patients with BRAFV600E+ systemic LCH) | Bilateral and symmetrical abnormalities (T2-w hyperintensity, T1-w isointensity or hypointensity) |
| Basal ganglia | — | Bilateral and symmetrical leukoencephalopathy-like abnormalities, or confluent lesions in a vascular pattern, with T2 hyperintensity and T1 hypointensity |
| Supratentorial white matter | Reactive gliosis and microglial activation decreased BRAFV600E+ cells compared with cerebellum/brainstem | Bilateral and symmetrical leukoencephalopathy-like abnormalities, or confluent lesions in a vascular pattern, with T2 hyperintensity and T1 hypointensity |
| Prominent, dilated perivascular spaces | ||
| Cerebral white matter | — | Bilateral and symmetrical punctate lesions in a vascular pattern. T2-w hyperintensity, and T1 iso- or hypointensity; variable contrast enhancement and mass effect |
Adapted from Yeh et al60 with permission.
LACS is a neurodegenerative syndrome of variable severity and course. The incidence of long-term neurodegeneration has been estimated to be between 1.9% and 11%,12,67 and it seems to be higher in patients with multisystem disease, diabetes insipidus, history of involvement of bones of the skull base and orbit,12,64,67,68 or BRAFV600E-mutated LCH.67 Of particular therapeutic relevance are the skull-based lesions (CNS-risk lesions), as this risk association has been considered an indication for the use of systemic therapy, rather than local control measures only.
The appearance of clinical and radiographic signs of LCH-ND can occur with the initial LCH diagnosis, although it commonly occurs years later.60,61,69 Symptoms may initially include tremors, abnormal reflexes, gait disturbance, motor spasticity, ataxia, dysarthria, dysphagia, behavioral changes, learning disorder, or psychiatric problems. Some patients develop a progressive cerebellar syndrome, with spastic tetraparesis, pseudobulbar palsy, and cognitive deterioration.60,61 Magnetic resonance imaging shows a characteristic infratentorial predilection, with symmetric abnormalities of the dentate nuclei and of the white matter of the cerebellum and pons (Figure 2; Table 4). Outside the infratentorial compartment, abnormalities of the basal ganglia, optic nerves, and tracts; dilatation of the Virchow-Robin spaces; or diffuse abnormalities of the hemispheric white matter consistent with leukoencephalopathy are also common.60,61 Serial imaging and neurocognitive evaluations are recommended when the disease is suspected.60,70
Whether CNS involvement with degeneration represents active disease or a radiologic scar remains undefined. Until recently, the only histologic study of LACS reported absence of CD1a+ histiocytes, an inflammatory collection of CD8+ lymphocytes with neuronal and axonal degeneration, and extensive myelin loss, supporting the view of a late consequence of an inflammatory phenomenon.71 However, a recent study supports hematopoietic origin of myeloid cells that share precursors with LCH lesion CD207+ cells.72 Clinical and radiological responses to BRAF inhibitors further support this view.73
Biology
Ontogeny and function of epidermal LCs
During development, a wave of LCs arises from yolk sac progenitors and fetal liver-derived monocytes and seeds the epidermis. This population is maintained locally, with tissue-resident precursors during steady state. However, when skin is injured or inflamed, monocyte-derived blood cells have the ability to migrate to epidermis and differentiate into LC-like cells.74-76 Migration of activated LC from epidermis to draining lymph node is dependent on C-C motif chemokine receptor 7 (CCR7).77 In the lymph node, LCs present antigen and activate T cells (Figure 4A).
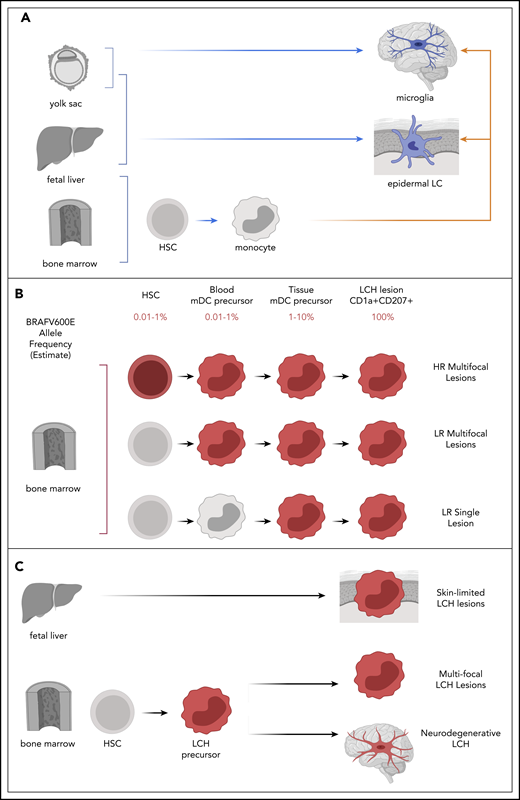
Models of LCH pathogenesis. (A) Ontogeny of physiologic epidermal LCs and microglia may inform mechanisms of LCH pathogenesis. Physiologic epidermal LCs arise from yolk sac and fetal liver; microglia arise from yolk sac (blue arrows). Subsequently, epidermal LC and microglia may be replace monocytes derived from bone marrow after activation or injury (orange arrows). (B) The variant allele frequency (BRAFV600E or other MAPK mutation) is found in very rare population in myeloid precursors in bone marrow and peripheral blood in patients with HR LCH and some with LR multifocal LCH. The Misguided Model of LCH Pathogenesis proposes that extent of disease is defined by the state of differentiation at which an activating somatic MAPK pathway gene mutation arises (red). (C) Lack of detectable PBMCs with BRAFV600E and self-resolving course support potential fetal liver origin, where identification of PBMCs with BRAFV600E and perivascular BRAF-V600E+ cells at sites of neurodegeneration support potential for hematopoietic clone (red) in LCH-ND. HSC, hematopoietic stem cell; HR, high risk; LR, low risk; mDC, myeloid dendritic cell.
Models of LCH pathogenesis. (A) Ontogeny of physiologic epidermal LCs and microglia may inform mechanisms of LCH pathogenesis. Physiologic epidermal LCs arise from yolk sac and fetal liver; microglia arise from yolk sac (blue arrows). Subsequently, epidermal LC and microglia may be replace monocytes derived from bone marrow after activation or injury (orange arrows). (B) The variant allele frequency (BRAFV600E or other MAPK mutation) is found in very rare population in myeloid precursors in bone marrow and peripheral blood in patients with HR LCH and some with LR multifocal LCH. The Misguided Model of LCH Pathogenesis proposes that extent of disease is defined by the state of differentiation at which an activating somatic MAPK pathway gene mutation arises (red). (C) Lack of detectable PBMCs with BRAFV600E and self-resolving course support potential fetal liver origin, where identification of PBMCs with BRAFV600E and perivascular BRAF-V600E+ cells at sites of neurodegeneration support potential for hematopoietic clone (red) in LCH-ND. HSC, hematopoietic stem cell; HR, high risk; LR, low risk; mDC, myeloid dendritic cell.
The evolving identity of LCH
Before LCH was histologically defined, cases of children with unusual constellations of symptoms were described. In the mid-1900s, pathologists noted the common histology of those disease presentations and hypothesized these conditions to represent a common disease entity.78,79 Lichtenstein proposed the nomenclature of Histiocytosis X, reflecting the uncertain cell of origin. In the 1960s, the Birbeck granules were identified and were thought to be exclusively associated with epidermal LCs (Figure 1). When Nezelof and colleagues identified Birbeck granules in Histiocytosis X lesion cells, Histiocytosis X was reframed as LC histiocytosis.80
Inflammation and LCH
Histologic similarities between LCH cells and epidermal LCs set the stage for a long-standing debate about LCH as a disorder of pathologic activation of epidermal LCs vs neoplastic transformation.81 The physiologic function of dendritic cells is to interact with and activate T cells, and LCH lesions are characterized by a robust immune infiltrate, although mechanisms driving inflammation are not well understood. The pathologic CD207+ dendritic cells (LCs) constitute a median of 8% of LCH lesion cells.82 The remainder of the lesion is composed of inflammatory infiltrate, including a significant population of T cells (enriched for activated CD4+ regulatory suppressor T cells) and abundant inflammatory cytokines.83-85 The LCs of an LCH lesion express high levels of the programmed cell death ligand 1, and infiltrating T cells express the programmed cell death protein.86
Recurrent somatic activating MAPK pathway gene mutations in LCH
In a long-standing cancer vs inflammation debate, nonrandom X-inactivation of CD1a+ cells supported clonality of LCH lesion LCs.87,88 However, LCs are not hyperproliferative within the lesion,83,84,89 and no gross genetic alterations were reported.90 With improved sequencing technologies, Rollins and colleagues analyzed CD1a+ cells isolated from LCH lesion biopsies and identified recurrent BRAFV600E mutations in more than 50% of the cases.91 Several groups subsequently found that BRAFV600E or alternative activating MAPK pathway gene mutations are nearly universal in LCH, including other BRAF mutations and mutations in MAP2K1 (encoding MEK1; Figure 5).92-96
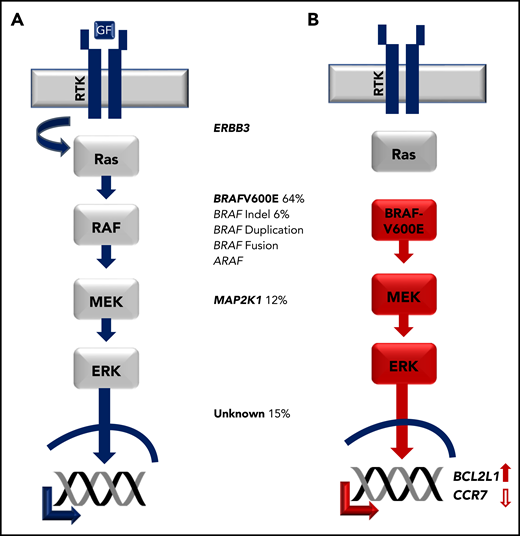
Activating MAPK pathway mutations in LCH. (A) Physiologic MAPK signaling transduces extracellular signal through receptor tyrosine kinase (RTK), which activates RAS, then RAF, then MEK, then ERK proteins, which in turn regulate cell-specific nuclear targets and gene transcription programs. (B) Activating mutations in LCH such as BRAFV600E drive constitutive ERK activation and downstream transcriptional targets including increased transcription of BCL2L1 and decreased CCR7.
Activating MAPK pathway mutations in LCH. (A) Physiologic MAPK signaling transduces extracellular signal through receptor tyrosine kinase (RTK), which activates RAS, then RAF, then MEK, then ERK proteins, which in turn regulate cell-specific nuclear targets and gene transcription programs. (B) Activating mutations in LCH such as BRAFV600E drive constitutive ERK activation and downstream transcriptional targets including increased transcription of BCL2L1 and decreased CCR7.
Origins of LCH lesion LCs
The unique phenotype of CD1a+CD207+ histiocytes made the epidermal LCs a likely culprit in LCH. However, gene expression comparing epidermal LCs with LCH lesion LCs identified a signature consistent with more immature myeloid precursors in the LCH cells.84 Further, multiple dendritic cell lineages beyond epidermal LCs have the capacity to express Langerin with potential for wide tissue distribution.76 Together, these observations supported potential for a hematopoietic origin and tissue distribution of precursor cells beyond the epidermis.
The discovery of somatic BRAFV600E mutation provided a molecular tag for lineage tracing to test the hypothesis that LCH could arise from hematopoietic precursors. Analysis of peripheral blood mononuclear cells (PBMCs) identified a very small (<1%) but consistent presence of BRAFV600E+ in myeloid cells (CD11c+ myeloid dendritic cell precursors and CD14+ monocytes) in patients with high-risk LCH with BRAFV600E+ lesions. Similarly, in bone marrow aspirates, BRAFV600E could be identified in CD34+ hematopoietic stem cells of many patients with high-risk LCH, ∼50% of which were reported as histologically normal. In contrast, BRAFV600E is typically absent from PBMCs of patients with active single lesion LCH and rarely identified in PBMCs of patients with multifocal low-risk LCH.97 These observations support the “misguided myeloid differentiation model” of LCH ontogeny, in which extent of disease is defined by differentiation of the cell in which activating somatic MAPK gene mutation (eg, BRAFV600E) arises (Figure 4B).
Patterns of skin LCH in infants and LCH-ND may inform pathogenic pathways in LCH. In an institutional series, BRAFV600E was frequently identified in PBMCs from children with multisystem LCH and skin lesions; however, BRAFV600E+ PBMCs were not typically detected in infants with skin-limited disease.98 This pattern could be consistent with skin-limited LCH in infants arising from a yolk sac or liver precursor during the wave of epidermal LC seeding that does not contribute to hematopoiesis. Multisystem LCH with BRAFV600E+ would be more consistent with a hematopoietic precursor (Figure 4C).
As discussed earlier, a recent series identified BRAFV600E+ cells in brain biopsies of patients with LCH-ND. Remarkably, a quantitative analysis of sections from whole-brain autopsy identified regions of brain stem and cerebellum with more than 10% BRAFV600E+ cells. In addition, BRAFV600E+ cells were identified in PBMCs in patients with LCH-ND who had no other systemic findings of active LCH, whereas BRAFV600E was not identified in patients cured from systemic LCH without LCH-ND. BRAFV600E+ cells concentrated around blood vessels and lack the phenotype of P2RY12+ microglia that arise during embryonic development.72 These observations support a model in which a hematopoietic clone could contribute to microglia-like cells that drive neurodegeneration (Figure 4C). The possibility of embryonic origin of LCH-ND is supported by a mouse model in which a yolk sac erythromyeloid precursor contributes to an isolated neurodegenerative process.99
Beyond BRAF
The MAPK pathway transduces extracellular signals that regulate transcriptional programs of cell growth, differentiation, and survival. The MAPK pathway is the most common dysregulated pathway in cancer, and BRAFV600E is identified in ∼8% of all cancers. It is infrequently associated with hematologic malignancies with the notable exception of hairy cell leukemia, and ERK activation affects myeloid cell differentiation and maturation.100,101 Cellular context likely plays a major role on the effect of MAPK pathway activation. In mice with enforced expression of BRAFV600E in CD11c cells, MAPK activation abrogates CCR7 expression in skin myeloid dendritic cells, which is required for their activation and migration to draining lymph nodes. Similarly, CCR7 expression is absent in CD207+ LCH lesion LC, but is rescued with MAPK inhibition. In addition, the BCL21 (encoding BCLXL), which inhibits apoptosis, is upregulated in myeloid dendritic cells with increased MAPK activation. Therefore, MAPK activation contributes to LCH pathogenesis by trapping cells at sites of lesions, where they accumulate and resist cell death.89
Do specific mutations matter? In vitro studies demonstrate differential ERK activation from different MAPK gene mutations, which might explain some differences in clinical presentation and outcomes. Increased rates of resistance to front-line chemotherapy, relapse, and neurodegeneration have been described in patients with BRAFV600E.72,97,67,102 Identifying mutation-specific pathogenic mechanisms may inform opportunities for clinical risk stratification and precision therapy.
Treatment of LCH
The difficulties in developing effective therapies for LCH are linked to the deficiencies in the understanding of its pathogenesis. Patients are now stratified into different risk categories based on the disease extent and the degree of organ dysfunction; patients with single-system disease confined to a single site usually require only local therapy or observation, whereas patients with more extensive disease require systemic therapy.23,27 These advances in risk-adapted treatment have resulted in better characterization of the natural history of the disease and an overall improvement in outcomes. Population-based studies have documented a significant increase in survival for patients with disseminated LCH, although these improvements appear to have favored children over adults, with 5-year relative survival rates of 90% vs 70%, respectively.6,8 Despite these improvements in survival, disease reactivations occur in approximately one-third of the patients,24,65,103 and their prevention has become one of the major objectives of current clinical trials. The current standard of care for front-line therapy of patients with multifocal LCH or unifocal disease in CNS-risk sites is vinblastine/prednisone for 1 year, with the potential addition of mercaptopurine for high-risk LCH.66 Robust data to guide treatment after first and subsequent treatment failures are lacking. LCH responds to increasing doses of nucleoside analogs with efficacy in other myeloid malignancies (cytarabine, cladribine, and clofarabine).104-106 For patients with low-risk disease recurrence, including patients with reactivation of single-system or multifocal bone disease or multisystem disease without risk organ involvement, less toxic regimens have proven to be effective, including oral 6-mercaptopurine and methotrexate,107 indomethacin,108 bisphosphonates,109 and hydroxyurea.110 The international LCH-IV protocol (ClinicalTrials.gov identifier: NCT02205762) represents a comprehensive effort to address the most relevant clinical and therapeutic challenges, including the management of upfront and relapsed LCH and the treatment of CNS disease.
Early-phase trials in adults with LCH and Erdheim-Chester disease support near-universal responses in patients treated with MAPK pathway inhibitors.50,111-114 Two recently reported retrospective pediatric series also report high response rates in cohorts including children with high-risk LCH, patients with multiple previous treatment failures, and patients with LCH-ND.73,115 However, rapid reactivations occur in the majority of patients after discontinuation of therapy, and reintroduction of the BRAF inhibitor is usually effective.115 Prospective trials will be required to determine optimal duration of therapy and potential for combination with other targeted or cytotoxic therapies.
A comprehensive review of historical and current clinical trials is detailed in the supplemental Materials, available on the Blood Web site.
Special treatment considerations
Treatment of adult LCH
Treatment of LCH in adults follows similar guidelines to those recommended for children, with some modifications.51,116 The more severe skin manifestations have shown to respond well to phototherapy,117 low-dose methotrexate,118 and thalidomide or lenalidomide.38 For patients requiring systemic therapy, vinblastine-based regimens remain quite effective in the adult population, with similar outcomes to children.119 However, given the diminished tolerance of adults to corticosteroids and vinblastine, treatment with cytarabine or cladribine is generally preferred,51,116,120 although BRAF inhibitors are being increasingly used in this population.50,121,122
For adults with pulmonary LCH, smoking cessation is critical for stabilization and improvement of symptoms, and a trial of observation after discontinuing smoking is recommended. For patients with severe or progressive disease, and for patients with multisystem disease, systemic therapy is recommended. Corticosteroids, either inhaled or systemic, have not shown to be of benefit. The most recommended treatment is cladribine, although the use of BRAF or MEK inhibitors in eligible patients should also be considered.56
Treatment of LCH CNS disease
There are no standard guidelines for treatment of LCH CNS disease. For tumorous lesions and new-onset diabetes insipidus, treatment with a standard LCH regimen is indicated; vinblastine and prednisone or single-agent cladribine have been shown to be effective.123,124 Treatment of LCH-ND is less defined. Improvement in the neurological condition has been reported with the use of cytarabine,125 intravenous immunoglobulins,126 rituximab,127 infliximab,128 and cis-retinoic acid.129 More recently, the documentation of diffuse perivascular infiltration by BRAFV600E cells in biopsies of patients with LCH-ND has provided a strong rationale for the use of targeted therapies72 ; responses to BRAF inhibitors have been documented in 12 of 13 patients.73
Late effects
Up to 50% of survivors have at least 1 permanent consequence.12,24,65 Long-term effects have been reported to be more frequent among patients with multisystem disease and patients with multiple reactivations.12,65 The most commonly reported late effects are diabetes insipidus and orthopedic abnormalities, which may occur in up to or slightly above 20% of patients, followed by growth retardation, hearing loss, and neurodegeneration in approximately 10% of the patients, and biliary cirrhosis, and respiratory insufficiency in less than 5% of patients.12,24,65 Of particular relevance is the neurodegenerative syndrome that usually occurs years after the original diagnosis, and which has been discussed here and extensively reviewed elsewhere.60,61
Future perspectives
Further research in the biology of LCH and its correlation with clinical presentation and outcomes will be required for better refinement of treatment of the disease and its complications, such as LCH-ND. Although the universal activation of the MAPK pathway provides a strong rationale for the use of pathway inhibitors, their role, including indications, optimal duration of treatment, and combination with standard chemotherapy, needs to be investigated.
The online version of this article contains a data supplement.
Acknowledgments
This work has been supported in part by a grant from the St. Baldrick’s Foundation for the North American Consortium for Histiocytosis (CRG and CEA), a Translational Research Program grant from the Leukemia and Lymphoma Society (CEA and CRG), and the American Lebanese Syrian Associated Charities (ALSAC).
Authorship
Contribution: C.R.-G. and C.E.A. jointly wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Carlos Rodriguez-Galindo, Department of Global Pediatric Medicine, St. Jude Children’s Research Hospital, 262 Danny Thomas Pl MS-721, Memphis TN 38105; e-mail: carlos.rodriguez-galindo@stjude.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal