


Abstract
Erdheim-Chester disease (ECD) is characterized by the infiltration of tissues by foamy CD68+CD1a− histiocytes, with 1500 known cases since 1930. Mutations activating the MAPK pathway are found in more than 80% of patients with ECD, mainly the BRAFV600E activating mutation in 57% to 70% of cases, followed by MAP2K1 in close to 20%. The discovery of BRAF mutations and of other MAP kinase pathway alterations, as well as the co-occurrence of ECD with LCH in 15% of patients with ECD, led to the 2016 revision of the classification of histiocytoses in which LCH and ECD belong to the “L” group. Both conditions are considered inflammatory myeloid neoplasms. Ten percent of ECD cases are associated with myeloproliferative neoplasms and/or myelodysplastic syndromes. Some of the most striking signs of ECD are the long bone involvement (80%-95%), as well as the hairy kidney appearance on computed tomography scan (63%), the coated aorta (40%), and the right atrium pseudo-tumoral infiltration (36%). Central nervous system involvement is a strong prognostic factor and independent predictor of death. Interferon-α seems to be the best initial treatment of ECD. Since 2012, more than 200 patients worldwide with multisystem or refractory ECD have benefitted from highly effective therapy with BRAF and MEK inhibitors. Targeted therapies have an overall, robust, and reproducible efficacy in ECD, with no acquired resistance to date, but their use may be best reserved for the most severe manifestations of the disease, as they may be associated with serious adverse effects and as-yet-unknown long-term consequences.
Introduction
Lipoid granulomatosis was first described by Erdheim and Chester in 1930.1 By July 2019, more than 1500 cases of this disease, now called Erdheim-Chester disease (ECD) after its discoverers, had been reported worldwide (including both published cases and patients from the ECD Global Alliance registry). The number of known patients has increased spectacularly during the last 15 years, mostly because of increased awareness and improved diagnostic acumen.
In 2010, BRAFV600E mutation was discovered in a landmark study of patients with Langerhans cell histiocytosis (LCH).2 Two years later, this mutation was shown to be recurrent and to occur at a similar frequency in histiocytes from ECD lesions.3 Several other kinase mutations or rearrangements of the RAS-RAF-MEK-ERK pathway were subsequently described,4,5 shedding light on the pathogenesis of histiocytosis and paving the way for treatments targeting BRAF and, a few years later, MEK in ECD. Such treatments have been used since 2012 and have proved effective against ECD.6-9 The discovery of BRAF mutations, and of other MAP kinase pathway alterations, led to the 2016 revision of the histiocytoses classification.10 ECD now belongs to the “L” group of this new classification, as well as LCH.
This review focuses on recent molecular discoveries and the clinical presentation, typical signs, and current standard of care for ECD, including the use of targeted therapies, now reported in more than 200 patients worldwide.
Clinical manifestations
The natural course of ECD is difficult to appreciate, mostly because in recent years, diagnosis almost invariably leads to treatment. However, in most cases, it seems to follow a smoldering course. When untreated, it can prove fatal, particularly in multisystem disease. Unlike LCH lesions, ECD lesions accumulate in affected organs and tissues, and spontaneous regression is rare.
ECD mostly affects adults (mean age at diagnosis, 55 years), with a male preponderance (sex ratio, 3:1).11-16 Childhood ECD is rare (<20 reported cases)17 and usually spares the heart. In 2006, a preliminary study reported a time between symptom onset and diagnosis of between several months and 25 years.18 The time to diagnosis has since decreased, undoubtedly because of greater awareness of the disease. Mean time from symptoms onset to diagnosis in our series of 261 patients seen at Pitié-Salpêtrière hospital (May 2019) was 2.7 years (median, 2 years; standard deviation, 10.9 years).
ECD can affect almost all systems and organs, and is frequently multisystem (Table 1).
ECD main organs involvement in the current series from Pitié-Salpêtrière Hospital (n = 261) and the National Institutes of Health cohort (n = 60) compared with the review from the literature
| Organ infiltration, clinical and mutational findings | Pitié-Salpêtrière (current series; n = 261) | Estrada-Veras et al (National Institutes of Health; n = 60)12 | Cives et al (review of literature; n = 448)15 |
|---|---|---|---|
| Long bone (metabolic, bone scintigraphy or PET), % | 79 | 95 | 74 |
| Bone pain, % | 38 | — | — |
| Cardiac involvement, % | 51 | — | 19 |
| Right atrium pseudotumor, % | 36 | 37 | 11 |
| Coronary infiltration, % | 23 | — | — |
| Pericardial involvement, % | 29 | 8 | 9 |
| Vascular involvement, % | 61 | — | 16 |
| Coated aorta, % | 40 | 62 | — |
| Xanthelasma, % | 22 | 33 | 27 |
| Diabetes insipidus, % | 25 | 47 | 48 |
| CNS involvement, % | 37 | 38 | 25-50 |
| Cerebellar involvement, % | 16 | — | — |
| Retro-orbital involvement, % | 20 | 27 | 35 |
| Lung involvement, % | 36 | 52 | 18 |
| Retroperitoneal involvement (hairy kidney aspect), % | 63 | 65 | 35 |
| Hydronephrosis, % | 27 | — | 6 |
| Organ infiltration, clinical and mutational findings | Pitié-Salpêtrière (current series; n = 261) | Estrada-Veras et al (National Institutes of Health; n = 60)12 | Cives et al (review of literature; n = 448)15 |
|---|---|---|---|
| Long bone (metabolic, bone scintigraphy or PET), % | 79 | 95 | 74 |
| Bone pain, % | 38 | — | — |
| Cardiac involvement, % | 51 | — | 19 |
| Right atrium pseudotumor, % | 36 | 37 | 11 |
| Coronary infiltration, % | 23 | — | — |
| Pericardial involvement, % | 29 | 8 | 9 |
| Vascular involvement, % | 61 | — | 16 |
| Coated aorta, % | 40 | 62 | — |
| Xanthelasma, % | 22 | 33 | 27 |
| Diabetes insipidus, % | 25 | 47 | 48 |
| CNS involvement, % | 37 | 38 | 25-50 |
| Cerebellar involvement, % | 16 | — | — |
| Retro-orbital involvement, % | 20 | 27 | 35 |
| Lung involvement, % | 36 | 52 | 18 |
| Retroperitoneal involvement (hairy kidney aspect), % | 63 | 65 | 35 |
| Hydronephrosis, % | 27 | — | 6 |
The first manifestation observed differs considerably between patients. Any of the many clinical signs, such as bone pain, xanthelasma, exophthalmos, tamponade, ataxia, constitutional symptoms, or sinusitis, may herald the disease. The most frequent first sign to appear during the course of the disease is almost certainly diabetes insipidus “of unknown origin” (16%), which may occur several decades before ECD is diagnosed,11 followed by neurological manifestations (14%).
Main clinical signs and involvements
Bone manifestations
The most frequent manifestation of ECD is long-bone osteosclerosis (Figure 1), which is observed in 80% to 95% of patients,11,12 and is symptomatic in 38%. It may cause mild leg bone pain,12 sometimes requiring nonsteroidal anti-inflammatory drugs or opioid treatment. Long-bone involvement is a hallmark of the disease.13 It may be detected by radiological imaging (X rays in the past, computed tomography [CT] scans and magnetic resonance imaging [MRI] more recently), or bone scintigraphy (99Technetium [99Tc]), revealing increases in radiotracer uptake by the distal ends of the femurs and the proximal and distal tibia. The latter have now largely been replaced by positron emission tomography-CT (PET-CT) showing avid FDG uptake in the legs detected in about 95% of patients. 18FDG-PET scans are particularly informative for assessments of ECD activity, in particular for therapeutic responses.19
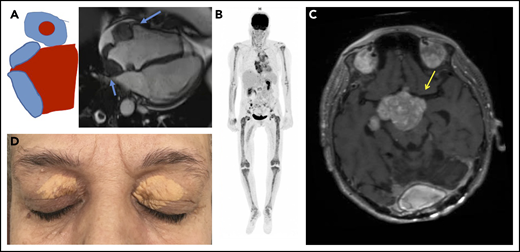
Typical clinical and imaging features of ECD. (A) Mass of the right atrium and of the atrioventricular sulcus (blue arrows). (B) Hypermetabolism of long bones, aorta, and right atrium in 18 fluorodeoxyglucose PET. (C) Pseudotumoral infiltration of brain (MRI, T1 weighted image with gadolinium), yellow arrow. (D) Typical xanthelasma palpebrarum
Typical clinical and imaging features of ECD. (A) Mass of the right atrium and of the atrioventricular sulcus (blue arrows). (B) Hypermetabolism of long bones, aorta, and right atrium in 18 fluorodeoxyglucose PET. (C) Pseudotumoral infiltration of brain (MRI, T1 weighted image with gadolinium), yellow arrow. (D) Typical xanthelasma palpebrarum
Cardiovascular manifestations
Progress in radiological imaging has greatly facilitated the detection of cardiovascular involvement. CT scan may reveal the sheathing of whole aorta (“coated aorta”), a classic feature of ECD (40% of our 261 cases). Peri-aortic infiltration may also affect either the thoracic or the abdominal portion of the aorta, and may extend to the main branches of the aorta.11,12,20 It is usually asymptomatic, and is not associated with dilation, dissection, or aneurysm. The infiltration of coronary arteries is seen in 23% of patients,11 and can be complicated by coronary stenosis and myocardial infarction.20 Pericardial disease is frequent (29%). It may present as pericarditis, pericardial effusion, or cardiac tamponade, and may be fatal.20
Right atrium (RA) pseudotumor, another classical feature of ECD, can be observed by dedicated MRI in 36% of patients.11,12,20 Dedicated cardiac MRI is the most appropriate type of imaging for assessments of cardiac manifestations of ECD, whereas aortic CT scans should be preferred for evaluations of peri-aortic sheathing.21 A recent retrospective study of cardiac imaging in 205 patients with ECD reported abnormal results for 101 patients (49%), including 72 with an impaired right ventricular atrioventricular sulcus (72%) and 65 with an impaired RA enclosure (64%).22
Urologic, nephrologic, and retroperitoneal infiltration
Retroperitoneal fibrosis is observed in a third of patients with ECD, sometimes complicated by bilateral hydronephrosis (27%), potentially requiring ureteral stenting.23 Pelvic ureter involvement has never been described, and the inferior vena cava is rarely affected. In ECD, fibrosis entirely encircles the aorta; in contrast, the posterior wall of the aorta is rarely affected in idiopathic retroperitoneal fibrosis.20 It should be noted that sheathing is observed not only around the aorta and the renal capsule but also often around the proximal portion of the ureters, and this is the cause of ureteral obstruction in most patients. In rare cases, the renal/perirenal lesions may be misdiagnosed as xanthogranulomatous pyelonephritis, with patients even undergoing bilateral nephrectomy in some cases.24 Abdominal CT scans reveal fat infiltration around the kidneys (“hairy kidneys”) in 63% of patients.11,12
Pulmonary manifestations
Lung involvement is seen on thoracic CT scans in 30% to 50% of cases.11,12,25 Infiltration of the pleura and lung parenchyma may be observed, with an interstitial lung disease-like pattern, including frequent smooth interlobular septal thickening or, more rarely, micronodules, ground-glass opacities, parenchymal condensation, thickening of interlobar fissure, and even occasional cysts, which are more typical of LCH.26 Of note, pleural involvement (predominantly on the right side) may be symptomatic and difficult to diagnose, and can be seen in 41% of patients.26
Many of the radiological patterns described here have been observed in our most recent cohort (36%), but very few patients display persistent pulmonary symptoms, such as respiratory failure (fewer than 10 patients), with little change to pulmonary functional test results generally observed. Nevertheless, it is important to check for this specific involvement, which has been identified as an independent predictor of poor prognosis in multivariate analysis.11
Endocrine manifestations
Patients with ECD frequently present endocrinopathies. Diabetes insipidus is found in 25% of patients, and is often the first manifestation of ECD,27 but it is not specific to this condition and is detected at a similar frequency in patients with LCH. In 91% of patients with full anterior pituitary evaluation, anterior pituitary dysfunctions, such as growth hormone deficiency (79%), hyperprolactinemia (44%), follicle-stimulating hormone and luteinizing hormone deficiencies (22%), thyrotropin deficiency (10%), and adrenocorticotropic hormone deficiency (3%), are detected. Dedicated MRI reveals infiltration of the pituitary and stalk in 24% of patients.27 Testicular insufficiency is found in 53% of patients, with 29% displaying sonographic testicular infiltration, mostly bilateral.27 CT shows adrenal infiltration in 39% of patients, but adrenal insufficiency is detected only very rarely.27 As in LCH, diabetes insipidus, when present, is almost always irreversible, although radiological infiltration of the pituitary may decrease on effective treatment.
Cutaneous manifestations
Xanthelasma are the most frequent cutaneous manifestation of ECD, occurring in 22% of patients,11,12,28 and classically affecting the eyelids or periorbital spaces. Reticular dermis involvement is more frequent than in classic xanthelasma palpebrarum, and the ECD-xanthelasma–like lesion contain larger numbers of multinucleated or Touton cells, with less extensive fibrosis.28 Nonspecific patches or papulonodular lesions, predominantly affecting the legs, back, and/or trunk, may also be observed.28 Infiltrations of the vulva and clitoris are observed much less frequently,1 and are less typical than in LCH.
Nervous system, facial, and orbital manifestations
As in LCH, neurological involvement in ECD may have a tumoral or degenerative presentation. Many different central nervous system (CNS) manifestations are observed in ECD. The most frequent neurological signs are cerebellar and pyramidal syndromes (41% and 45% of cases, respectively), but seizures, headaches, sensory disturbances, neuropsychiatric signs, cognitive impairment, cranial nerve palsy, and asymptomatic lesions have also been reported.29 Degenerative involvement of the cerebellum is observed in 17% of patients and is very difficult to treat. On MRI, CNS manifestations may appear as enhancing masses or high signal density. Imaging shows diffuse thickening or dural masses indicative of meningeal involvement in 23% of patients.30 CNS involvement can lead to severe disability, and survival analysis has identified this factor as an independent predictor of death (hazard ratio, 2.51).31 Intracranial vascular infiltration, particularly in the basilar artery vessels, is also observed in some patients with ECD and can lead to ischemic stroke. Five patients of our current series have an important infiltration around intracranial arteries, especially the basilar trunk. This peri-arterial intracranial encasement is far less frequent than the coated aorta. CSF findings are usually normal in neurological ECD.
A recent retrospective analysis of all CNS manifestations (both clinical and imaging) in 253 patients with ECD followed-up in our hospital showed CNS involvement in 97 patients (38%). These patients were significantly younger at diagnosis (mean age, 55.5 years) and at symptom onset (mean age, 50.5 years), and were more likely to have xanthelasma (34%) and diabetes insipidus (36%). Median survival was significantly shorter in patients with CNS involvement than in those without such involvement (124 vs 146 months). A central review of 74 CNS MRI scans identified 3 nonexclusive patterns: tumoral in 66%, degenerative in 50%, and vascular in 18%.
A quarter of all patients develop exophthalmos as a result of infiltration of the retro-orbital soft tissues, and this condition is often bilateral.11,12,32 Sinus infiltration is also frequent, particularly for maxillary and sphenoid sinuses (47%), as opposed to the ethmoidal and frontal sinuses (17%). PET-CT may reveal hypermetabolism of the sinuses, an element supporting ECD diagnosis.
Other manifestations
As in LCH, gum and tooth involvement may occasionally be observed in patients with ECD, and appropriate care is required for periodontal disease and missing teeth in particular. The gut and liver are involved much less frequently than in LCH, but exudative enteropathy and secondary sclerosing cholangitis have been reported.33 In rare cases, some patients may suffer from mesenteric angina as a result of sheathing of mesenteric superior artery or of celiac trunk.20
Cases of macrophage activation syndrome have been published, and infiltrations of other organs (thyroid, breast,34 pancreas, lymph nodes) have been described, but are mostly rare. Autopsy procedures have also demonstrated cases of thyroid and lymph node infiltrations.35 In some cases, patients may also experience fever, which may be specific of the disease and its activity.
Laboratory abnormalities
Serum C-reactive protein levels are high in 80% of cases.18 Systemic and local inflammation seem to be the main driver of organ damage. Assays for 23 cytokines on serum samples from 37 patients with ECD revealed high interferon (IFN)-α, interleukin (IL)-1/IL1-RA, IL-6, IL-12, and MCP-1 levels, indicating strong, systemic immune activation36 and evidence of an association between ECD and systemic Th-1-oriented immune disturbance. Immunohistochemical analyses in 3 patients showed histiocyte recruitment and accumulation in lesions to be governed by a complex network of cytokines and chemokines.37 A study of the spontaneous and stimulated production of cytokines by mononuclear cells in biopsy fragments from a single patient showed that TNF-α was produced after stimulation, whereas IL-6 and IL-8 were secreted spontaneously; IL-8 served as a chemoattractant for polymorphonuclear cells and monocytes.38 Levels of chemokine ligand 18, which has been implicated in the induction and progression of fibrosis, were 3 to 4 times higher in a series of 20 patients with ECD than in control individuals, and were correlated with disease severity.39
Diagnosis
ECD is diagnosed by characteristic radiological findings and appropriate histology, sufficient to exclude other mimics such as malignancy and autoimmune disease.13 The lesions are generally infiltrated with foamy histiocytes and surrounded by fibrosis or xanthogranulomatosis. Touton giant cells are frequently, but not always, present. ECD histiocytes are CD68+ CD163+ FXIIIa+ and CD1a− (corresponding to “non-LCH” histiocytes). The organ lesions are generated by a proliferative mechanism, although the evolution is usually very slow, as Ki67 levels are low in most tissue histiocytes.
A biopsy is required to establish the diagnosis of ECD, and a CT-guided biopsy of perirenal infiltration or skin lesions, such as xanthelasmas, is recommended when these involvements are present.
The following criteria were used for ECD diagnosis both in our 1996 and 2004 surveys20,40 :
Characteristic histological findings (Figure 2): foamy histiocyte infiltration of polymorphic granuloma and fibrosis or xanthogranulomatosis, with immunostaining positive for CD68 and negative for CD1a.
Characteristic skeletal abnormalities: bilateral symmetric cortical osteosclerosis of the diaphyseal and metaphyseal parts of the long bones on X ray, and/or symmetric abnormally intense labeling of the distal ends of the long bones of the legs, and in some cases, arms, on 99Tc bone scintigraphy. However, PET-CT has largely superseded bone CT scans during the last decade as a tool for detecting long-bone infiltration.
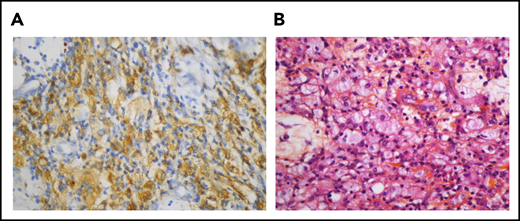
Pericardial localization of ECD. (A) The histiocytes are positive for CD68 (immunostaining with KP1 clone, original magnification, ×400). (B) Pericardial infiltrate made up of histiocytes, some of them with foamy cytoplasm, admixed with a few lymphocytes (hematein-eosin-saffron, original magnification, ×400)
Pericardial localization of ECD. (A) The histiocytes are positive for CD68 (immunostaining with KP1 clone, original magnification, ×400). (B) Pericardial infiltrate made up of histiocytes, some of them with foamy cytoplasm, admixed with a few lymphocytes (hematein-eosin-saffron, original magnification, ×400)
These criteria remain useful, but it has become clear, with much larger case series, that some patients with ECD may not necessarily present typical long-bone involvement (either morphologically or metabolically). In such patients (<5% of all patients with ECD), diagnosis requires robust histological results to detect molecular alterations (see "Mutational landscape of ECD") and/or other typical clinical signs of involvement (eg, hairy kidney, coated aorta, pseudotumoral infiltration of the RA and sinus infiltration). In particular, the detection of a BRAFV600E mutation or of another MAPK pathway alteration has become the gold standard and is of key importance to diagnose ECD in the absence of typical features (see Table 2).
Revised diagnostic criteria proposed for ECD, based on recent molecular findings
| Clinical or morphological findings |
| a) Symmetric diaphyseal and metaphyseal osteosclerosis in the legs, best visualized on PET and/or |
| b) Other typical ECD manifestations: perirenal infiltration (hairy kidney) best visualized on CT scan; periaortic sheathing (coated aorta) best seen on CT scan; right atrial pseudo-tumor, best visualized on cardiac MRI; xanthelasma; exophthalmos; osteosclerosis of the facial sinuses |
| Histopathological findings |
| Typical foamy or lipid-laden histiocytes admixed with fibrosis sometimes with Touton giant cells |
| Immunohistochemical staining positive for CD68 or CD163 and negative for CD1a |
| Molecular findings |
| Identification in histiocytosis tissue sample of |
| a) BRAFV600E mutation or |
| b) Activating mutation in the BRAF gene other than BRAFV600E or along the MAPKinase pathway such as KRAS, NRAS, MAP2K1, ARAF, MAP3K1 (nonexhaustive list) or |
| c) Gene fusion activating the MAPKinase pathway or |
| d) Activating mutation in CSF1R |
| Clinical or morphological findings |
| a) Symmetric diaphyseal and metaphyseal osteosclerosis in the legs, best visualized on PET and/or |
| b) Other typical ECD manifestations: perirenal infiltration (hairy kidney) best visualized on CT scan; periaortic sheathing (coated aorta) best seen on CT scan; right atrial pseudo-tumor, best visualized on cardiac MRI; xanthelasma; exophthalmos; osteosclerosis of the facial sinuses |
| Histopathological findings |
| Typical foamy or lipid-laden histiocytes admixed with fibrosis sometimes with Touton giant cells |
| Immunohistochemical staining positive for CD68 or CD163 and negative for CD1a |
| Molecular findings |
| Identification in histiocytosis tissue sample of |
| a) BRAFV600E mutation or |
| b) Activating mutation in the BRAF gene other than BRAFV600E or along the MAPKinase pathway such as KRAS, NRAS, MAP2K1, ARAF, MAP3K1 (nonexhaustive list) or |
| c) Gene fusion activating the MAPKinase pathway or |
| d) Activating mutation in CSF1R |
ECD diagnosis should be made when criterion 1 is associated with criterion A and/or B.
Differential diagnosis of ECD
Thickening of the aorta wall, extending to the main branches of the aorta, may be observed in Takayasu arteritis, which typically affects young women. However, radiological findings differ between Takayasu arteritis and patients with ECD: the entire wall (adventitia, media, and intima) is affected in Takayasu arteritis, whereas the adventitial and periadventitial periaortic spaces, but not the wall itself, are affected in patients with ECD.20
Radiologic characteristics can also help in distinguishing between ECD and mediastinal and retroperitoneal fibrosis (RPF). Idiopathic RPF is typically not circumferential, infiltrating the anterior and lateral sides of the aorta, but sparing the posterior side. Inferior vena cava (stenosed or occluded) or pelvic ureter involvement may be observed in RPF, but not in ECD. Extravascular images of bilateral infiltration of the perirenal space (hairy kidneys), for example, can be useful for differentiating between ECD and RPF.
Immunoglobulin G4 (IgG4)-related disease (IgG4-RD) is a multisystem condition that may be confused with ECD. It is characterized histologically by dense lymphoplasmacytic infiltrates rich in IgG4+ plasma cells, abundant fibrosis with a storiform pattern, lymphoid follicles, tissue eosinophilia, and obliterative phlebitis. Serum IgG4 levels are high in about half the patients with IgG4-RD,41 but may also be elevated in ECD. The typical manifestations of IgG4-RD are lymphadenitis, sclerosing pancreato-cholangitis, lacrimal and salivary gland involvements, and tubulo-interstitial nephritis, which are usually absent in ECD. ECD-related RPF generally affects the perirenal areas, renal pelvis, and proximal ureters (typical hairy kidneys). In contrast, IgG4-RD RPF usually develops around the antero-lateral sides of the abdominal aorta and iliac arteries (although atypical cases of isolated ureteral involvement have been described). ECD-related RPF obstructs the proximal ureters, whereas IgG4RD-related RPF encases the distal part of the ureters and determines medial ureteral deviation.42
Of note, other histiocytoses (such as Rosai-Dorfman-Destombes disease [RDD]) should also be evoked in the differential diagnosis.
Mutational landscape of ECD
The RAS-RAF-MEK-ERK cellular signaling pathway plays a major role in solid tumors and hematological malignancies. The BRAFV600E activating mutation is present in several human tumors43 at various frequencies (eg, 40%-60% in melanoma, 100% in hairy cell leukemia, 5% in papillary thyroid cancer). It activates the RAS-ERK pathway independent of RAS, through several different cell functions including proliferation, apoptosis, angiogenesis, migration, and survival.43 Using a mass spectrometry analysis targeting cancer genes (OncoMap), in 2010, Barrett Rollins’s group was able to detect the recurrent presence of BRAFV600E variant in 57% of 61 LCH archived samples.2 The high prevalence of this authentic oncogenic driver unambiguously classified LCH as a clonal disease,44 albeit with a marked inflammatory component. A review of histological data from 127 patients with various types of histiocytoses resulted in the detection of BRAFV600E mutations by pyrosequencing in 93 patients. BRAFV600E mutations were detected in 13 (54%) of 24 ECD, and 11 (38%) of 29 patients with LCH, but not in patients with any other type of histiocytosis tested (eg, RDD, xanthoma disseminatum). Immunohistochemical analyses of ECD tissue samples with a BRAFV600E-selective antibody confirmed the expression of the mutated BRAF protein in typical foamy histiocytes and Touton giant cells, but not in lymphocytes, fibroblasts, or endothelial cells.3
Immunostaining for phosphorylated extracellular signal-regulated kinase is positive in almost all ECD tissue samples.5 This strongly suggested a role for other activating mutations of the MAPK pathway: a combined whole-exome and transcriptome analysis of large cohorts of tissue samples taken from patients with ECD found recurrent mutations of MAP2K1, ARAF, NRAS, and KRAS, and translocations of BRAF, ALK, and NTRK1. MAP2K1 mutations were detected in almost 30% of cases, and 27% of patients had KRAS or NRAS mutations.4,10 Very rare BRAF deletions, MAP3K1amp, ARAF mutations, and RET fusions have also been characterized. Activating PIK3CA mutations have been found in 11% of patients; the MAPK pathway is, therefore, not the only signaling pathway affected in this disease.5 These mutations activate the PI3K–AKT pathway, which can also be induced via the MAPK pathway. Moreover, CD68+ histiocytes express cytoplasmic phosphorylated mechanistic targets of rapamycin and p70S6K.45 Highly sensitive methods, such as digital droplet polymerase chain reaction, are required to detect mutations in histiocytoses, including ECD in particular. Less sensitive techniques would fail to pick up 25% of the BRAF mutations, because of the low variant allele frequency (≤5%).46
We found a correlation between BRAF status and the most severe sites of ECD involvement, which has an importance in terms of treatment (see "Treatments"). The presence of the BRAFV600E mutation is significantly associated with RA pseudotumor (57% vs 9% for BRAFWT patients; P < .0001)11 and a higher risk for cardiac and aortic infiltration (odds ratio per 1 standard deviation increase in BRAFV600E status, 4.92).47 BRAF mutation is also associated with pericardial involvement.22 Most of the 97 patients of our cohort (38%) with CNS involvement also had BRAFV600E mutations (77% of cases).
Overlap histiocytoses
Histiocytoses are heterogeneous diseases, and associations of several different types of histiocytosis within the same patient, or even the same biopsy specimen, have been observed. This situation is particularly frequent for ECD and LCH. Indeed, 15.3% of the 261 patients followed at our center presented such associations: ECD and LCH in 33 cases, ECD and RDD in 4 cases, and even LCH, ECD and RDD in a single case. Although evoked when facing certain radiologic or clinical signs, the definition of overlap histiocytoses is histologic. The high frequency of ECD/LCH overlap was highlighted by Hervier et al,48 who reported 23 such cases: ECD followed (n = 12) LCH, or was diagnosed with it (n = 11), but never preceded LCH. The phenotypes of patients with mixed histiocytosis were heterogeneous, but closer to isolated ECD than isolated LCH. BRAFV600E mutations were found on both ECD and LCH biopsies for 8 patients, indicating that the association of LCH and ECD is not fortuitous, and may be linked to BRAF. ECD was recently found in association with RDD in 13 cases (12 men), 7 of which displayed testes involvement, with MAP2K1 mutation in 9 cases.49
Association of ECD and myeloid neoplasms
Two tertiary care centers retrospectively reviewed the tissue samples and medical records of 189 patients with ECD or mixed histiocytosis. They found that 10.1% had overlapping myeloid neoplasms50 : chronic myelomonocytic leukemia in half the cases and myeloproliferative neoplasm or myelodysplastic syndrome in most of the others. JAK2V617F was the most frequent mutation in myeloid neoplasms, followed by NRAS, TET2, ASXL1, and U2AF1 mutations. Moreover, a sequential analysis of colonies derived from single CD34+ cells from a patient with TET2- and SRSF2-mutated chronic myelomonocytic leukemia associated with BRAFV600E-mutated ECD detected colonies with BRAF and TET2 mutations, with the TET2 mutations systematically preceding the BRAF mutations.51
Treatments
Many therapeutic regimens, including steroids, cytotoxic agents, vinca alkaloids (by analogy with LCH treatment), and double-autologous hematopoietic stem cell transplantation, were described in limited case series before 2005.52 The recent advances in targeted therapies have led to the abandonment of stem cell transplantation for this indication. IFN-α-based regimens have been shown to improve survival,31 but the most promising advances since 2012 have been the demonstrations of efficacy for BRAF inhibitors and MEK inhibitors.6,9 IFN-α treatment is necessarily long-term and is associated with a number of adverse effects, such as depression and fatigue, which affect half the patients.53 The 5-year survival rate of our 261 patients with ECD in May 2019 was 79%. In contrast, life expectancy was much lower in 2004, with up to 60% of patients dying within 3 years of diagnosis. IFN-α is still the best initial choice for ECD treatment. We usually initiate treatment with PEGylated forms, which are better tolerated than standard forms. The therapeutic response (by PET-CT and conventional imaging) of IFN-based regimens is usually much slower and more partial compared with that seen with targeted therapies. Assessments should be made every 6 months, after an initial point after 3 months when using targeted therapies.
In cases of mild or nonsevere ECD (eg, without CNS and/or heart involvement) and contraindications to or adverse effects to IFN-α, the alternative treatments include anakinra,54 infliximab,55 and sirolimus + steroids.38
Vemurafenib, a BRAF inhibitor, increases survival in patients with metastatic melanomas carrying the BRAFV600E mutation.56 In 2012, we treated 3 patients with multisystem refractory ECD carrying the BRAFV600E mutation with vemurafenib. Two patients also had skin or lymph node LCH, and all 3 had highly robust responses.6 We subsequently treated 5 other patients with this drug, which was effective in the long term.7,8 Similar responses were obtained in 26 patients from the VE-BASKET trial (22 with ECD, 4 with LCH) from the Memorial Sloan-Kettering Cancer Center.57 We have continued to treat ECD and mixed histiocytosis with BRAF and MEK inhibitors. In 2017, we reported the results obtained with targeted therapies (BRAF and MEK inhibitors) in 54 patients with ECD, focusing on follow-up, adverse effects, and the results of treatment interruption from a clinical trial (the LOVE study, Long-term Outcome after Vemurafenib).8 Confirmed relapses occurred after a median of 6 months in 75% of the patients who stopped treatment. Treatment was then recommenced in 10 patients, leading to remission. The most frequent adverse effects when treated with vemurafenib are skin complications, ranging from pilar keratosis and photosensitivity to spinocellular carcinoma, melanoma, sarcoidosis-like disease, and DRESS (drug reaction with eosinophilia and systemic symptoms), but also arthralgias, pancreatitis, and QT prolongation. Its toxicity is usually dose limited and is the main reason for using lower doses in ECD than in the melanoma trials. Recently, myelodysplastic syndrome and pancreatic adenocarcinoma have also been reported. The other BRAF inhibitor, dabrafenib, seems to have a better safety profile with equivalent efficacy. The most frequent adverse effects of the MEK inhibitor cobimetinib are nausea, acneiform rash, and rhabdomyolysis. Altogether, these results led to approval by the US Food and Drug Administration of vemurafenib for BRAF-mutated ECD in November 2017.
The robust efficacy of MEK inhibition in 3 patients with multisystem and refractory ECD treated with cobimetinib was reported in 2016.58 In 2019, Diamond et al showed cobimetinib to be effective in 12 patients with ECD (overall response rate, 89%).9 Sustained responses were obtained, with no acquired resistance to date. Treatment efficacy was not affected by genotype, and responses were observed in patients with ARAF, BRAF, RAF1, NRAS, KRAS, MEK1 (also known as MAP2K1), and MEK2 (also known as MAP2K2) mutations.
Almost 200 adult patients with ECD or ECD and LCH (mixed histiocytosis) have been treated since 2012 with targeted therapies (BRAF inhibitors and/or MEK inhibitors). An awareness of targeted therapies adverse effects is therefore important, and although we believe such treatments should only be kept for those patients with the most severe forms of ECD (eg, CNS and/or heart involvements), other centers in North America will likely advocate targeted therapies at a lower threshold of disease burden/earlier in lines of therapy. Targeted therapies have nevertheless an overall robust efficacy in ECD, with no acquired resistance to date, and we truly hope all patients with severe ECD worldwide will get access to such therapies in the near future.
Authorship
Contribution: J.H., F.C.-A., and Z.A. designed and wrote this review; F.C.-A. conducted the statistical analysis and made Figure 2; and all authors critically reviewed and approved the final version of this manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Julien Haroche, Service de Médecine Interne 2, Maladies Auto-Immunes et Systémiques, Institut E3M, Hôpital Pitié-Salpêtrière, 47-83 de l’Hôpital, 75651 Paris CEDEX 13, France; e-mail: julien.haroche@aphp.fr.
