


Abstract
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV)–driven B-cell lymphoproliferative disease (LPD). This disease is hypothesized to result from defective immune surveillance of EBV, with most patients showing evidence of immune dysfunction, despite no known primary immunodeficiency. Pathologically, LYG is graded by the number and density of EBV+ atypical B cells, and other characteristic findings include an angioinvasive/angiodestructive reactive T-cell infiltrate and various degrees of necrosis. Clinically, LYG universally involves the lungs with other common extranodal sites, including skin, central nervous system, liver, and kidneys. Nodal and/or bone marrow involvement is extremely rare and, if present, suggests an alternative diagnosis. Treatment selection is based on histologic grade and underlying pathobiology with low-grade disease hypothesized to be immune-dependent and typically polyclonal and high-grade disease to be immune-independent and typically oligoclonal or monoclonal. Methods of augmenting the immune response to EBV in low-grade LYG include treatment with interferon-α2b, whereas high-grade disease requires immunochemotherapy. Given the underlying defective immune surveillance of EBV, patients with high-grade disease may have a recurrence in the form of low-grade disease after immunochemotherapy, and those with low-grade disease may progress to high-grade disease after immune modulation, which can be effectively managed with crossover treatment. In patients with primary refractory disease or in those with multiple relapses, hematopoietic stem cell transplantation may be considered, but its efficacy is not well established. This review discusses the pathogenesis of LYG and highlights distinct histopathologic and clinical features that distinguish this disorder from other EBV+ B-cell LPDs and lymphomas. Treatment options, including immune modulation and combination immunochemotherapy, are discussed.
Introduction
Lymphomatoid granulomatosis (LYG) is a rare Epstein-Barr virus (EBV)–associated B-cell lymphoproliferative disorder (LPD) that has unique histopathologic and clinical features that distinguish it from other EBV+ B-cell LPDs and lymphomas. This disorder was first described in 1972 by Liebow et al1 and, because of the predominance of T cells on pathologic examination, was initially thought to be a T-cell disorder.2,3 Subsequently, LYG was determined to be a B-cell LPD secondary to EBV,4,5 with a prominent angiocentric T-cell infiltrate.6,7 In 2001, LYG was incorporated into the World Health Organization classification of Tumors of Hematopoietic and Lymphoid Tissues and remains in the 2016 revision as a distinct mature B-cell neoplasm.8,9 As EBV is associated with several malignant and nonmalignant B-cell disorders (Table 1), distinct clinicopathologic findings can be used to distinguish LYG from other EBV-associated lesions. Herein, we discuss the pathogenesis of LYG and highlight distinct histopathologic and clinical features that distinguish this disease from other EBV+ B-LPDs and lymphomas. Treatment options for LYG are also discussed, with an emphasis on immune modulation and immunochemotherapy.
EBV-associated B-cell LPDs
| Infectious mononucleosis |
| Posttransplant and other iatrogenic B-cell LPDs |
| Monomorphic, polymorphic, plasmacytic, or HL-like variants |
| Lymphomatoid granulomatosis |
| EBV+ DLBCL associated with chronic inflammation |
| Fibrin-associated DLBCL |
| Pyothorax-associated lymphoma |
| EBV+ mucocutaneous ulcer |
| Hodgkin’s lymphoma |
| Subset of classic and HIV-associated |
| Mainly mixed cellularity and lymphocyte depleted |
| Primary effusion lymphoma (HHV-8 and EBV) |
| Germinotropic B-LPD (HHV-8 and EBV) |
| Plasmablastic lymphoma |
| Primary CNS lymphoma |
| Immunocompromised patients (e.g. HIV) |
| EBV+ DLBCL, NOS |
| Burkitt lymphoma |
| All endemic and a subset of sporadic and HIV associated |
| Infectious mononucleosis |
| Posttransplant and other iatrogenic B-cell LPDs |
| Monomorphic, polymorphic, plasmacytic, or HL-like variants |
| Lymphomatoid granulomatosis |
| EBV+ DLBCL associated with chronic inflammation |
| Fibrin-associated DLBCL |
| Pyothorax-associated lymphoma |
| EBV+ mucocutaneous ulcer |
| Hodgkin’s lymphoma |
| Subset of classic and HIV-associated |
| Mainly mixed cellularity and lymphocyte depleted |
| Primary effusion lymphoma (HHV-8 and EBV) |
| Germinotropic B-LPD (HHV-8 and EBV) |
| Plasmablastic lymphoma |
| Primary CNS lymphoma |
| Immunocompromised patients (e.g. HIV) |
| EBV+ DLBCL, NOS |
| Burkitt lymphoma |
| All endemic and a subset of sporadic and HIV associated |
HL, Hodgkin lymphoma; HHV-8, human herpesvirus 8; NOS, not otherwise specified.
Disease pathogenesis
LYG has a complex relationship with the host’s underlying immune function and defective immune surveillance of EBV-infected B cells, particularly a functional defect in CD8+ cytotoxic T cells, is hypothesized to lead to disease development.10,11 All LYG patients treated at the National Cancer Institute (NCI) demonstrated moderately to severely decreased baseline CD3+ T cells, with a greater decrease in CD8+ compared with CD4+ cells, and a less prominent reduction in NK and B-cell subsets.10,12 Also, these patients all showed serologic evidence of prior EBV exposure; however, none had clinical or serologic evidence of acute EBV infection at presentation.10 Taken together, these results suggest that the immunologic deficits are likely preexistent and that a quantitative and/or qualitative defect in mainly CD8+ cytotoxic T cells may be a prerequisite for disease.
Histologic grading and pathologic findings
Histologic grading of LYG is based on the number and density of EBV+ atypical B cells, and accurate grading is essential to better predict disease course and guide therapy. In a large pathologic series from the NCI, approximately half of the cases were classified as low-grade (grades 1 and 2) and high-grade (grade 3) disease.13 In accordance with the World Health Organization classification, grade 1 lesions were associated with rare atypical medium-to-large lymphoid cells, whereas a greater number of large atypical cells, occasionally reminiscent of Hodgkin/Reed-Sternberg cells, were frequently seen in grade 2 and 3 lesions (Figure 1).13 EBV, as assessed by in situ hybridization for EBV-encoded RNA (EBER), was seen in approximately half of grade 1 lesions with a paucity of EBER+ cells (<5 cells per high-power field [hpf]), making these lesions more difficult to diagnose than higher grade disease. In contrast, near universal expression of EBER was seen in grade 2 (5-20 and occasionally up to 50 cells per high-power field) and grade 3 (>50 and often >100 cells per high power field) lesions (Figure 1).
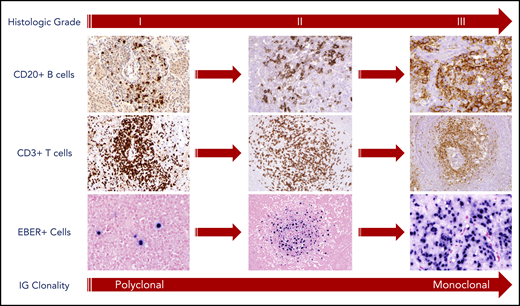
Pathologic findings and histologic grading of LYG. Histologic grading of LYG is based on the number and density of EBV+ atypical B cells and degree of coagulative necrosis. Low-grade (grades 1 and 2) disease is characterized by sparse EBER+ atypical B cells, as well as focal or absent coagulative necrosis. An increased number and size of EBER+ atypical B cells are seen in high-grade (grade 3) disease with often-extensive coagulative necrosis. A reactive angiocentric and angiodestructive CD3+ T-cell infiltrate is another characteristic pathologic feature across all grades of the disease. An increased frequency of monoclonality by molecular analysis is seen with higher grades of disease, likely representative of the progressive transformation of EBV-infected B cells.
Pathologic findings and histologic grading of LYG. Histologic grading of LYG is based on the number and density of EBV+ atypical B cells and degree of coagulative necrosis. Low-grade (grades 1 and 2) disease is characterized by sparse EBER+ atypical B cells, as well as focal or absent coagulative necrosis. An increased number and size of EBER+ atypical B cells are seen in high-grade (grade 3) disease with often-extensive coagulative necrosis. A reactive angiocentric and angiodestructive CD3+ T-cell infiltrate is another characteristic pathologic feature across all grades of the disease. An increased frequency of monoclonality by molecular analysis is seen with higher grades of disease, likely representative of the progressive transformation of EBV-infected B cells.
Baseline histologic grade correlates with underlying disease biology and clonality in LYG. In our NCI series, immunoglobulin gene (IG) rearrangement studies using polymerase chain reaction (PCR) showed various degrees of clonality across histologic grades, with low-grade LYG infrequently clonal by IG PCR (8% and 50% clonally rearranged in grades 1 and 2, respectively) with a trend toward increased clonality in grade 3 lesions (69% clonally rearranged).13 This shift in clonality from polyclonal to monoclonal disease seen with increased histologic grade most likely represents the selective transformation of EBV-infected B-cell clones (Figure 1).
LYG is also characterized pathologically by its polymorphous angiocentric and angiodestructive infiltrate, leading to varying degrees of coagulative necrosis.13 The inflammatory infiltrates in LYG consists mainly of small lymphocytes, plasma cells, and histiocytes and are associated with varying levels of angioinvasion and angiodestruction affecting small to large caliber vessels (Figure 1).13 Most of the angiocentric lymphocytes are CD3+ T cells, with CD4+ cells representing the predominant subtype in most cases. Coagulative necrosis may be seen in LYG of all histological grades, but is less common and generally is focal only in grade 1 lesions, in contrast to the often-extensive necrosis with CD20+ “ghost cells” seen in grade 2 and 3 lesions.13
The pathogenesis of tissue necrosis and vascular damage in LYG and other EBV+ B-LPDs is hypothesized to be both chemokine-mediated and directly related to invasion of inflammatory cells responding to EBV infection. Prior studies have implicated induction of the CXC chemokines CXCL9 and CXCL10 (formerly known as the monokine induced by IFN-γ [Mig] and IFN-γ–inducible protein 10 [IP-10]).14 CXCL9 and CXCL10 localize to the reactive cells in the viable tissue surrounding areas of necrosis, suggesting that cells in the tumor microenvironment and not the malignant cells themselves are the principal source of chemokines, resulting in vascular damage and necrosis.14 Thus, the host immune response to EBV is a principal cause of the vasculitic changes that are such a typical feature of the disease.
Cutaneous lesions in LYG are characterized pathologically by a dense dermal or subcutaneous lymphohistiocytic infiltrate with nonnecrotizing granulomatous inflammation, focal necrosis, and only sparse EBER+ cells seen in approximately one-quarter of cases.13 In our NCI series, higher EBV positivity was seen in patients with papular or nodular lesions compared with those with plaque lesions (38% vs 0%, respectively), and similar to pulmonary findings, monoclonality by IG PCR correlated with higher histologic grade and greater proportion of EBV+ B cells.15 Given the frequent subcutaneous panniculitis and nonnecrotizing granulomatous inflammation and infrequent EBV positivity seen in the skin, the cutaneous manifestations may also be the result of a systemic, rather than local, effect of EBV. For this reason, skin lesions alone are inadequate to establish a definitive diagnosis of LYG.
Clinical characteristics
LYG typically presents in middle-aged adults in the fourth to sixth decades of life and has an approximate 2:1 male predominance (Table 2).4,12,13,16-19 The disease is universally extranodal; thus conventional staging with the Ann Arbor staging system is uninformative, given that, by definition, all cases are considered stage IV. In LYG patients at the NCI, universal lung involvement was seen with the next most common sites of disease including the central nervous system (CNS; 40%), skin (34%), kidney (19%), and liver (17%) (Table 2; Figures 2 and 3).12,13 Lymph node and bone marrow involvement are extremely rare in LYG and thus, if found on diagnostic work-up, should raise suspicion for an alternative diagnosis.
Characteristics of LYG patients from an interim analysis of the prospective NCI study
| Characteristic | All patients (N = 70) |
|---|---|
| Median age (range), y | 46.2 (14.9-67) |
| Male sex, n (%) | 45 (64) |
| Histologic grade, n (%) | |
| I | 19 (27) |
| II | 24 (34) |
| III | 27 (39) |
| Disease sites, n (%) | |
| Lung | 70 (100) |
| CNS | 28 (40) |
| Skin | 24 (34) |
| Kidney | 13 (19) |
| Liver | 12 (17) |
| Spleen | 7 (10) |
| LN | 4 (6) |
| Other | 13 (19) |
| Prior therapy, n (%) | |
| None | 23 (33) |
| Steroids | 32 (46) |
| Chemotherapy with/without rituximab | 21 (30) |
| Rituximab | 13 (19) |
| Characteristic | All patients (N = 70) |
|---|---|
| Median age (range), y | 46.2 (14.9-67) |
| Male sex, n (%) | 45 (64) |
| Histologic grade, n (%) | |
| I | 19 (27) |
| II | 24 (34) |
| III | 27 (39) |
| Disease sites, n (%) | |
| Lung | 70 (100) |
| CNS | 28 (40) |
| Skin | 24 (34) |
| Kidney | 13 (19) |
| Liver | 12 (17) |
| Spleen | 7 (10) |
| LN | 4 (6) |
| Other | 13 (19) |
| Prior therapy, n (%) | |
| None | 23 (33) |
| Steroids | 32 (46) |
| Chemotherapy with/without rituximab | 21 (30) |
| Rituximab | 13 (19) |
LN, lymph node.
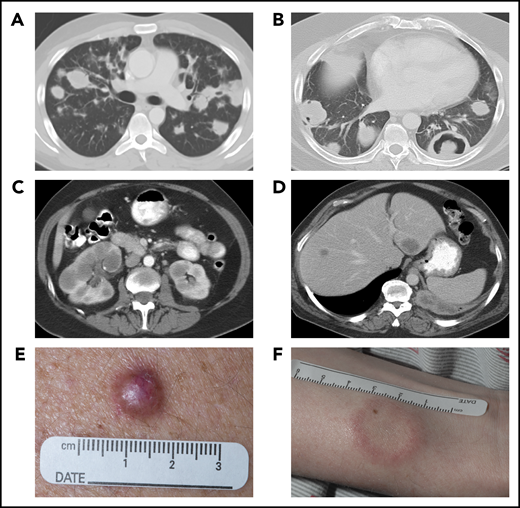
Systemic imaging and cutaneous manifestations of LYG. Computed tomography (CT) of the chest in a 25-year-old man (A) showed multiple, bilateral nodular masses affecting the mid to lower lung fields and in a 43-year-old man (B) showed central necrosis and cavitation in some of the nodular pulmonary lesions. CT of the abdomen in a 57-year-old woman (C) showed multiple, bilateral renal masses and in a 52-year-old man (D) revealed 2 hypodense liver lesions. Photographs of the skin of a 56-year-old man (E), with an erythematous dermal papule without ulceration, and of a 29-year-old man (F), with a white plaquelike lesion with surrounding erythema.
Systemic imaging and cutaneous manifestations of LYG. Computed tomography (CT) of the chest in a 25-year-old man (A) showed multiple, bilateral nodular masses affecting the mid to lower lung fields and in a 43-year-old man (B) showed central necrosis and cavitation in some of the nodular pulmonary lesions. CT of the abdomen in a 57-year-old woman (C) showed multiple, bilateral renal masses and in a 52-year-old man (D) revealed 2 hypodense liver lesions. Photographs of the skin of a 56-year-old man (E), with an erythematous dermal papule without ulceration, and of a 29-year-old man (F), with a white plaquelike lesion with surrounding erythema.
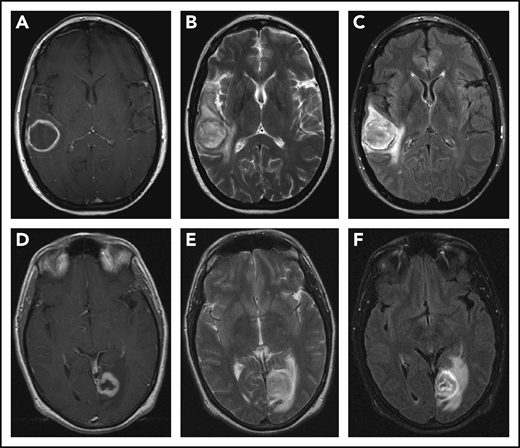
CNS imaging findings in LYG. T1-weighted postcontrast (A,D), T2-weighted (B,E), and fluid attenuation inversion recovery (C,F) sequences from MRI of the brain in a 43-year-old woman (A-C) with LYG demonstrates a large, enhancing right temporal lesion with central necrosis, and in a 39-year-old man (D-F) with LYG demonstrates a large, enhancing left posterior occipital lesion with central necrosis.
CNS imaging findings in LYG. T1-weighted postcontrast (A,D), T2-weighted (B,E), and fluid attenuation inversion recovery (C,F) sequences from MRI of the brain in a 43-year-old woman (A-C) with LYG demonstrates a large, enhancing right temporal lesion with central necrosis, and in a 39-year-old man (D-F) with LYG demonstrates a large, enhancing left posterior occipital lesion with central necrosis.
Laboratory findings in LYG are typically nonspecific and insufficient to lead to a diagnosis.1,4,20 Signs of immune dysfunction are usually present with alteration in immune subsets.20 Specifically, a greater decrease in CD3+ T cells is seen compared with B-cell or NK-cell subsets with a more pronounced decrease in CD8+ over CD4+ T cells.10,12 Serologic studies routinely show evidence of prior EBV exposure without findings of acute EBV infection (EBV IgG+ and IgM−), and low-level viremia by EBV DNA polymerase chain reaction (∼100 copies/mL) is frequently observed.10,12 Despite these frequent signs of immune dysfunction, typically no clear immunodeficient state can be identified by standard testing in most patients.
Despite universal lung involvement, approximately one- to two-thirds of patients present with overt respiratory symptoms, which trigger imaging, with the remaining patients identified incidentally by abnormal pulmonary imaging.16,20 Symptoms, when present, are variable and can range from cough and dyspnea to chest pain and may be accompanied by constitutional symptoms, such as fever, weight loss, malaise, and fatigue.16,20,21 Pulmonary imaging typically reveals multifocal bilateral nodular masses that predominantly affect the mid to lower lung fields (Figure 2A); however, occasional presentation with a solitary pulmonary nodule may be encountered.13,16,19 The nodular lesions can show evidence of central necrosis or cavitation (Figure 2B), both at initial presentation of disease and after therapy.
Approximately one-third of LYG patients have involvement of the skin with 2 main distinct patterns of cutaneous involvement.4,12,13,15,19 Multiple erythematous dermal papules and/or subcutaneous nodules, with or without ulceration, are most frequent (Figure 2E), followed less commonly by multiple indurated, erythematous to white plaquelike lesions (Figure 2F).15,22 Cutaneous lesions are frequently disseminated across the entire body and are less commonly seen only on the extremities, trunk, or head and neck.15,22 These lesions can occur at any stage of the disease process, with approximately one-third of patients having skin lesions at initial presentation.15 Cutaneous lesions rarely develop before pulmonary manifestations, however, and are somewhat nonspecific both histologically and clinically, resembling other causes of lobular panniculitis.
CNS involvement is seen in approximately one-third of LYG cases and demonstrates a variable spectrum of clinical symptoms and imaging findings.12,13,17,19 Neurologic symptoms depend on the site of CNS involvement and may include hearing loss, diplopia, dysarthria, hemiparesis, ataxia, and atonic bladder, and many but not all patients with abnormal radiographic findings on brain magnetic resonance imaging (MRI) have clinical symptoms.17 Focal intraparenchymal lesions are most common on MRI and are characterized by multiple foci of abnormal increased signal intensity on fluid attenuation inversion recovery and T2-weighted imaging (Figure 3).17 Abnormal punctate or linear foci of enhancement are frequently observed in these lesions, with enhancing lesions typically residing along medullary vessels, reflective of the angiocentricity of disease. Enhancing lesions can also evolve over time with up to one-third of heterogeneously enhancing intracranial masses evolving into a ringlike enhancing lesion with a nonenhancing center (Figure 3A,D).17 Abnormal enhancement of the cranial nerves and/or leptomeninges is also seen in LYG and can lead to cranial nerve palsy of the affected nerves.17 Less common imaging findings include enhancement and engorgement of the choroid plexus and/or thickening of the dura mater.
No clear association between disease grade and dissemination to the CNS has been observed, with similar rates of CNS involvement by imaging in patients with high- and low-grade disease.17 In addition, the sensitivity of CSF analysis for the diagnosis of CNS LYG is low, and only approximately one-third of patients with abnormal findings on baseline MRI have concurrent abnormalities in the CSF detected by cytology or flow cytometry.17 Abnormal CSF findings are most common in patients with cranial nerve and/or leptomeningeal involvement on imaging, indicating that CSF analysis may aid in confirmation of CNS involvement when imaging findings in these regions are subtle or inconclusive.17
Treatment
Given the rarity of LYG, there is no standard treatment consensus, and treatment has varied widely from observation to systemic corticosteroids, anti-CD20 monoclonal antibody therapy (rituximab), and chemotherapy. Several older retrospective studies suggest that no mode of therapy is satisfactory, with up to two-thirds of patients dying, many within the first year of diagnosis.4,18,20,22 Given the frequent polyclonal nature of low-grade disease and its association with EBV B-cell proliferation, grade 1 and 2 LYG was hypothesized to be immune dependent and therefore potentially treatable with agents that augment the immune response to EBV.10 Grade 3 LYG, however, is frequently monoclonal and was thus hypothesized to be relatively immune independent, similar to other EBV+ lymphomas, therefore requiring cytotoxic immunochemotherapy.
Based on this hypothesis, an on-going prospective treatment study of LYG was initiated at the NCI in 1994 to assess a treatment paradigm based on using disease pathobiology to guide therapy.12 Patients with low-grade LYG receive initial therapy with dose-escalated IFN-α2b, whereas patients with high-grade disease receive initial treatment with dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab (DA-EPOCH-R). Crossover therapy was allowed for emergence of alternate grade disease after initial treatment. An identical treatment paradigm was used for untreated patients, as well as those with relapsed/refractory disease. With the use of this pathobiologically based treatment approach, interim results have demonstrated improved outcomes compared with historic controls, with more than half of treated patients surviving at least 10 years with nearly 13 years of median follow-up.12
Low-grade LYG treatment
Given the relationship of LYG to immune function, standard lymphoma treatments, such as immunochemotherapy and/or steroids risk further worsening the immune surveillance of EBV and overall disease. Corticosteroids and/or single-agent chemotherapy have been used in prior studies to decrease or stabilize LYG lesions4,11,23-25 ; however, these measures typically fail to provide long-term disease control with a high rate of progression to high-grade disease or overt lymphoma (∼10% to 20%), typically resulting in death within 1 year.1,4,18,20,22 Occasional durable remissions can be achieved in LYG with rituximab26-30 ; however, as a monotherapy these responses are typically transient, with most patients progressing and requiring treatment with immune modulation and/or immunochemotherapy.30-33
Agents known to augment the immune response, such as IFN-α and intravenous gamma globulin, have been used in immunodeficient patients because of the direct antiviral and antiproliferative effect on EBV-infected B cells.34 In an early small series of patients with B-cell LPDs treated with the combination of IFN-α and intravenous gamma globulin, dramatic clinical responses were observed in all patients, suggesting that this is an effective treatment approach in polyclonal and monoclonal B-cell processes in immunodeficient patients.34 Favorable initial results were also demonstrated in a small number of LYG patients treated with IFN-α monotherapy, all of whom responded to therapy, with 3 of 4 patients achieving a complete response (CR) and remaining disease-free at a median of nearly 4 years.10
Observation/immunosuppression withdrawal
Although patients with iatrogenic immune dysfunction are at risk of LYG,35-39 these patients, as well as those with primary immunodeficiencies, are thought to have a disease process distinct from the typical LYG seen in otherwise immunocompetent individuals. Nonetheless, iatrogenic causes of immunosuppression should be reduced or discontinued in LYG if possible, and patients with low-grade disease may be observed for regression.36-39 Even in the absence of a reversible cause of immune dysfunction, some patients with low-grade LYG and a low disease burden may be eligible for an initial period of surveillance. A minority of these cases may show no progression or spontaneous remission4 ; however, most eventually require disease-directed therapy.
IFN-α
In the prospective NCI study, patients with low-grade LYG received primary therapy with IFN-α, initially at a dose of 7.5 million international units (MIU) 3 times weekly (TIW), which was escalated as tolerated, every 1 to 2 weeks and continued for 1 year past best response. Interim analysis of the 49 IFN-α–treated patients revealed a median dose and duration of IFN-α therapy of 20 MIU TIW and ∼8 months, respectively.12 Of the evaluable patients, approximately two-thirds experienced a response to therapy (overall response rate, 60%) with more than half achieving a CR.12 Similar responses to IFN-α, including CRs, were observed in patients with baseline CNS involvement, suggesting that this therapy is capable of penetrating the blood-brain barrier and eradicating CNS disease. Furthermore, these responses were often durable, with more than one-third of patients maintaining a response at 10 years, despite the 1 year of planned therapy (10-year progression-free survival [PFS] and overall survival [OS] of 37% and 64%, respectively).12 Of note, a subset of patients diagnosed initially with low-grade LYG progressed to high-grade disease within the first few months of IFN-α therapy. It is hypothesized that these cases harbored unidentified high-grade lesions before treatment that emerged during primary therapy for low-grade disease.
Other immunotherapies
Immune surveillance plays a key regulatory role in the development of malignancy, and EBV+ lymphomas are hypothesized to have acquired tolerogenic mechanisms that permit immune evasion.40,41 Upregulation of programmed death ligand 1 (PD-L1) is one mechanism by which abnormal cells can evade immune surveillance, and high levels of PD-L1 expression have been demonstrated in EBV+ B-LPDs.40,42-44 Blocking the interaction of programmed death 1 (PD-1) with its ligands, PD-L1 and PD-L2, has been shown to reverse the inactivation of tumor-specific effector T cells and to activate antitumor responses.45-47 PD-1 blockade may be an effective therapeutic strategy in LYG and other EBV+ B-LPDs. Ongoing clinical trials are assessing the efficacy of the PD-1 inhibitor nivolumab as a monotherapy (clinicaltrials.gov #NCT03258567) and in combination with other immunotherapies (#NCT03038672) in patients with EBV+ B-LPDs, including LYG and lymphoma. Furthermore, EBV-specific cytotoxic T-lymphocyte (CTL) infusions have shown durable remissions in patients with EBV+ B-LPDs and lymphomas, including 1 patient with LYG who achieved a durable CR for more than 4 years.48,49 Additional studies using CTLs and/or chimeric antigen receptor T-cell therapy are needed to better define the role of these cellular therapies in LYG.
High-grade LYG treatment
In contrast to low-grade LYG, high-grade disease is believed to represent an autonomous process that is less likely to respond to agents that augment the immune response to EBV. The treatment approach to high-grade disease has followed a paradigm similar to that of other aggressive lymphomas, such as EBV+ diffuse large B-cell lymphoma (DLBCL), mainly involving combination immunochemotherapy. Early treatment studies in LYG used a treatment approach modeled on Wegener’s granulomatosis, based on the hypothesis that LYG shared similar disease mechanisms associated with vasculitis. The first prospective study in LYG used a combination of low-dose oral cyclophosphamide and prednisone, with 7 (54%) of the 13 patients who received both therapies achieving CR with a mean remission duration of ∼5 years.20 Eight (53%) of 15 patients ultimately died, with 7 deaths related to evolution of disease into malignant lymphoma.20 Despite the early promising efficacy of single-agent chemotherapy with corticosteroids, more aggressive combined chemotherapeutic regimens were ultimately thought to be needed to better control disease and potentially prevent progression to overt lymphoma.
Combination immunochemotherapy
In the prospective NCI study, patients with high-grade LYG receive primary therapy with DA-EPOCH-R (dose-adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) every 3 weeks for up to 6 cycles of therapy. Interim results from the 18 primary DA-EPOCH-R–treated patients show this regimen to be highly active in high-grade disease, with more than three-quarters of evaluable patients achieving a response to therapy (overall response rate, 77%) with 41% CR.12 Approximately one-third of patients experienced a durable remission with a 5-year PFS and OS of 28% and 66%, respectively, with most of the remaining patients relapsing with low-grade disease.12 Similar findings were noted in another small retrospective study at Moffitt Cancer Center that evaluated 11 LYG patients (45% grade 3) treated with other rituximab-based therapies, mainly cyclophosphamide, doxorubicin, vincristine, and prednisone (R-CHOP).16 Response to therapy was achieved in approximately two-thirds of patients, with one-third attaining a CR; median PFS and OS were ∼1 and ∼2 years, respectively.16
Other combination immunochemotherapy regimens. as well as high-dose chemotherapy with autologous stem cell transplantation, have demonstrated efficacy in patients with LYG; however, these results are limited to anecdotal case reports and series.32,50-53 Given the low number of patients and the lack of comparative data, superiority among the various immunochemotherapy regimens cannot be determined, and routine use of high-dose chemotherapy/autologous stem cell transplantation as a front-line or adjuvant therapy cannot be recommended in LYG.
Targeted therapies
Targeted agents, such as histone deacetylase inhibitors (HDACis) and immunomodulatory drugs have been shown to induce EBV lytic-phase gene expression and to sensitize EBV+ lymphoma cells to cytotoxic effects of antiviral agents.54,55 HDACis and immunomodulatory drugs have demonstrated single-agent activity in aggressive B-cell lymphomas,56-58 and in an early clinical study, the HDACi nanatinostat in combination with ganciclovir, led to clinical responses in EBV+ B- and T-cell lymphomas.59 Further studies are needed using these targeted therapies in LYG to determine whether similar responses can occur in patients with this disorder.
Treatment of relapse/progression
Crossover treatment
Given defective immune surveillance of EBV in LYG, frequent relapse with low-grade disease or progression to high-grade disease occurs after immunochemotherapy or immune modulation in high- and low-grade disease, respectively. In the prospective NCI study, patients with progression after primary IFN-α therapy and those who progressed or failed to achieve CR after DA-EPOCH-R could cross over to alternative treatment. Based on interim results, 19 patients with LYG progressed to high-grade disease during or after treatment with IFN-α and received secondary DA-EPOCH-R.12 Approximately three-quarters of patients experienced a response to secondary therapy, with more than half achieving CR, including patients with disease refractory to primary IFN-α. Alternatively, secondary IFN-α therapy was given to 8 patients who progressed or relapsed with low-grade disease during or after DA-EPOCH-R.12 Secondary IFN-α was active in these patients as well with approximately three-quarters and two-thirds of patients achieving an overall response and CR, respectively. It is important for treating physicians to recognize the frequent emergence of different grades after primary therapy, as crossover treatment remains highly effective in these cases.
Hematopoietic stem cell transplantation
In patients with refractory LYG or multiple relapses, hematopoietic stem cell transplantation (HSCT) may eradicate recurrent disease. A small series of patients with multiple relapses of LYG treated with HSCT was reported by the European Group for Blood and Marrow Transplantation.60 Ten patients underwent a total of 9 autologous stem cell transplantations and/or 4 reduced-intensity allogeneic HSCT (allo-HSCT) with all patients having active disease at the time of transplantation. With a median follow-up of 5.1 years, 6 patients remained alive and disease free with 2 relapses and 4 deaths after transplantation.60 Despite the limitations of this small series and potential for selection bias, given the poor outcome of patients with multiple relapses or refractory disease, consideration should be given to HSCT in patients fit to undergo such therapy.
Differential diagnosis of LYG
Historical reports have noted the development of LYG in patients with immunodeficiency, including Wiskott-Aldrich syndrome and HIV,6,9,61,62 and notably, there is some degree of histological overlap between LYG and other forms of EBV-driven LPD. One key distinction between LYG and other EBV+ B-LPDs and lymphomas relates to the underlying pathophysiology of organ damage in these diseases. In most EBV+ B-LPD, direct expansion of EBV-infected B cells is the main contributor to the pathological damage incurred by tissues. In contrast, organ damage in LYG seems less directly related to EBV-infected B cells and may in part be mediated by the host’s immune response toward EBV, which produces the characteristic vasculitis that is such a constant feature of the disease.14 It is this aspect, which is characterized by an angiocentric T-cell infiltrate, that helps identify LYG as a separate entity. However, it is likely that both defective immune surveillance and an abnormal immune response toward EBV contribute to LYG pathogenesis.
EBV+ B-LPDs comprise a diverse group of LPDs (Table 1), and, in some instances, the differential diagnosis with LYG can be challenging, particularly when the process involves extranodal sites (Table 3). Most often in LYG, no clear immunodeficient state can be identified, which is in contrast to the known secondary immunodeficiency in polymorphic posttransplant lymphoproliferative disorder (PTLD) that occurs in the setting of solid organ or allogeneic bone marrow transplantation.13 In EBV+ DLBCL, immune senescence plays a major role, with most cases presenting in the elderly.41,63 Polymorphic PTLD can have a polymorphous infiltrate and necrosis on pathologic examination similar to LYG; however, the content of EBV+ cells is lower in LYG, particularly in low-grade lesions.13 Extranodal disease with involvement of the lung, CNS, and skin is common to both LYG and polymorphic PTLD; however, gastrointestinal tract involvement is more frequent in polymorphic PTLD, and the allograft itself is occasionally involved.64-67 Although EBV+ DLBCL may involve extranodal sites, this is less common than in LYG and more than half of EBV+ DLBCL cases present concurrently with nodal involvement.13,40,41,63 EBV viral load is often markedly elevated in both polymorphic PTLD and EBV+ DLBCL, which is in contrast to the low-level viremia typical of LYG.68,69 The overall histologic features in many of these conditions may show overlap, highlighting the importance of the clinical context.70,71 Although pathological lesions characteristic of LYG occur in the setting of congenital or acquired immunodeficiency, it is likely that such cases are distinct from classic LYG, requiring a modified designation in the future.
Comparison of LYG with polymorphic PTLD and EBV+ DLBCL
| LYG | Polymorphic PTLD | EBV+ DLBCL | |
|---|---|---|---|
| Clinical features | Immunodeficient state often unknown | Immunodeficient state known | Often immunocompetent, but immune senescence plays a role |
| Extranodal involvement frequent, most commonly lung, CNS, and skin | Extranodal involvement frequent, most commonly gastrointestinal, lung, CNS, kidney, heart and liver, including the allograft | Extranodal involvement less common and involving diverse sites | |
| Lymph node and/or bone marrow involvement extremely rare | Lymph node involvement uncommon and bone marrow disease rare | Lymph node involvement common and bone marrow disease uncommon | |
| Pathologic features | EBV+ atypical B cells in a T-cell rich background | EBV+ atypical B cells predominate | EBV+ atypical B cells predominate |
| Angiocentric and angioinvasive polymorphous infiltrate present | Polymorphous plasma cell-rich infiltrate present but typically not angiocentric or angioinvasive | EBV+ large B cells predominate | |
| Various degrees of coagulative necrosis | Coagulative necrosis may be present | Various degrees coagulative necrosis | |
| EBV viral load | Often low to negative | Often significantly elevated | Often significantly elevated |
| LYG | Polymorphic PTLD | EBV+ DLBCL | |
|---|---|---|---|
| Clinical features | Immunodeficient state often unknown | Immunodeficient state known | Often immunocompetent, but immune senescence plays a role |
| Extranodal involvement frequent, most commonly lung, CNS, and skin | Extranodal involvement frequent, most commonly gastrointestinal, lung, CNS, kidney, heart and liver, including the allograft | Extranodal involvement less common and involving diverse sites | |
| Lymph node and/or bone marrow involvement extremely rare | Lymph node involvement uncommon and bone marrow disease rare | Lymph node involvement common and bone marrow disease uncommon | |
| Pathologic features | EBV+ atypical B cells in a T-cell rich background | EBV+ atypical B cells predominate | EBV+ atypical B cells predominate |
| Angiocentric and angioinvasive polymorphous infiltrate present | Polymorphous plasma cell-rich infiltrate present but typically not angiocentric or angioinvasive | EBV+ large B cells predominate | |
| Various degrees of coagulative necrosis | Coagulative necrosis may be present | Various degrees coagulative necrosis | |
| EBV viral load | Often low to negative | Often significantly elevated | Often significantly elevated |
Conclusions
LYG is a rare EBV-associated LPD characterized pathologically by various numbers of EBV+ atypical B cells in addition to a reactive angioinvasive/angiodestructive T-cell infiltrate. Disease pathogenesis results from defective immune surveillance, mainly in CD8+ cytotoxic T cells, against the EBV virus and in most cases no clear immunodeficient state can be identified. Clinically, the disease universally involves the lungs, with other extranodal sites such as the CNS, skin, liver, and kidneys frequently involved. Lymph node and/or bone marrow involvement is extremely rare and, if present, suggests an alternative diagnosis. Treatment of LYG is dependent on histologic grade and underlying disease biology, and approximately one-third of patients can achieve durable remission with immune modulation and combination immunochemotherapy for low- and high-grade disease, respectively. Frequent progression to high-grade disease and relapse with low-grade disease occurs after immune modulation and immunochemotherapy, respectively, in low- and high-grade LYG, which can be effectively managed through crossover treatment. In refractory LYG or patients with multiple relapses, autologous stem cell transplantation and/or allo-HSCT may be considered, and, given the efficacy of EBV-specific CTLs and chimeric antigen receptor T-cell therapy in other lymphomas, cellular immunotherapies such as these are worth studying in refractory LYG.49,72,73
Acknowledgments
The authors thank Mark Roschewski for helping to write and review the manuscript.
Research funding for the prospective National Cancer Institute study was provided by the intramural program of the National Institutes of Health.
Authorship
Contribution: C.M. wrote the manuscript; E.S.J. provided pathologic imaging and helped write and review the manuscript; and W.H.W. helped write and review the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Wyndham H. Wilson, Lymphoid Malignancies Branch, CCR, NCI, Building 10, Room 4N115, 9000 Rockville Pike, National Institutes of Health, Bethesda, MD 20892; e-mail: wilsonw@mail.nih.gov.
