


Abstract
Systemic mastocytosis (SM) has greatly benefited from the broad application of precision medicine techniques to hematolymphoid neoplasms. Sensitive detection of the recurrent KIT D816V mutation and use of next-generation sequencing (NGS) panels to profile the genetic landscape of SM variants have been critical adjuncts to the diagnosis and subclassification of SM, and development of clinical-molecular prognostic scoring systems. Multilineage KIT involvement and multimutated clones are characteristic of advanced SM (advSM), especially SM with an associated hematologic neoplasm (AHN). A major challenge is how to integrate conventional markers of mast cell disease burden (percentage of bone marrow mast cell infiltration and serum tryptase levels) with molecular data (serial monitoring of both KIT D816V variant allele frequency and NGS panels) to lend more diagnostic and prognostic clarity to the heterogeneous clinical presentations and natural histories of advSM. The approval of the multikinase/KIT inhibitor midostaurin has validated the paradigm of KIT inhibition in advSM, and the efficacy and safety of second-generation agents, such as the switch-control inhibitor ripretinib (DCC-2618) and the D816V-selective inhibitor avapritinib (BLU-285) are being further defined in ongoing clinical trials. Looking forward, perhaps the most fruitful marriage of the advances in molecular genetics and treatment will be the design of adaptive basket trials that combine histopathology and genetic profiling to individualize treatment approaches for patients with diverse AHNs and relapsed/refractory SM.
Molecular genetics
Mutations in KIT
More than 90% of patients with systemic mastocytosis (SM) have a gain-of-function mutation in codon 816 of the receptor tyrosine kinase KIT, where a valine is substituted for an aspartate (KIT D816V).1 KIT D816V is located in exon 17 and renders KIT constitutively active and resistant to imatinib. Alternative KIT mutations in codon 816 (eg, D816A/F/H/I/N/T/Y) are uncommon and functionally equivalent to D816V. In addition to the tyrosine kinase domain (exons 17 and 18; eg, D820G or N822I/K), a majority of ∼30 different KIT mutations have been identified in the extracellular (exons 8-9), transmembrane (exon 10; eg, F522C) and juxtamembrane domains (exon 11; eg, V560G/I). These rare mutations are generally imatinib-sensitive.2
The low sensitivity of Sanger sequencing (∼10%-20%), as well as next-generation sequencing (NGS; ∼5%), may generate false-negative results in a significant proportion of patients. Highly sensitive polymerase chain reaction (PCR) assays on DNA or RNA (sensitivity, ∼0.01%-0.1%) allow the identification of KIT D816V in peripheral blood (PB) of nearly all genuinely KIT D816V+ patients.3 These assays, which can be used for the quantification of the KIT D816V variant allele frequency (VAF) using DNA4-6 or the expressed allele burden using RNA,7,8 have become important complementary tools for diagnosis, because the detection of KIT D816V serves as 1 minor diagnostic criterion for SM9 and defines which small molecules exert activity against this canonical KIT variant.
Multilineage involvement of KIT D816V
At least 60% to 80% of patients with advanced SM (advSM) present with signs of proliferation and/or dysplasia. If diagnostic criteria are met, a diagnosis of an associated hematologic neoplasm (AHN) can be made. AHNs are usually of myeloid origin, such as chronic myelomonocytic leukemia (CMML; most common), myelodysplastic syndrome (MDS), myeloproliferative neoplasm (MPN), MDS/MPN unclassifiable, chronic eosinophilic leukemia (CEL), or even acute myeloid leukemia (AML).9-11 Rarely, lymphoid neoplasms such as chronic lymphocytic leukemia or multiple myeloma are found with SM.
Although KIT D816V is often thought of as a mast cell (MC)–restricted mutation, Sotlar et al12 first reported the presence of KIT D816V in monocytic bone marrow (BM) infiltrates of patients with SM-CMML, supporting the hypothesis of coevolution of SM and CMML. Moreover, KIT D816V could be identified in variable myeloid subtypes of AHN (eg, CMML, 89%; MPN, 20%; AML, 30%).13 Using flow cytometry–sorted populations of BM cells in 113 patients, the Spanish Network on Mastocytosis (REMA) found multilineage involvement of the mutation in virtually all patients with aggressive SM (ASM) and in 20% to 30% of patients with indolent SM (ISM).14 In a long-term study of 145 patients, multilineage involvement of KIT D816V and elevated β2-microglobulin were the only independent prognostic factors for progression in ISM.15
AdvSM is a multimutated disease: impact of additional somatic mutations beyond KIT D816V
Sotlar et al16 first identified somatic mutations besides KIT D816V in SM patients. In 5 patients with associated primary myelofibrosis, they detected KIT D816V in all patients, and JAK2 V617F was found in the AHN component as well as in microdissected MCs in 4 of 5 patients. Conversely, KIT D816V was identified in MCs, microdissected granulocytes, and CD15+ cells in 2 of 5 patients. These data show that KIT D816V and JAK2 V617F coexist in the neoplastic cells of both disease components.
Additional somatic mutations (eg, TET2, SRSF2, ASXL1, EZH2, CBL, RUNX1, RAS) have been found in ∼90% of advSM patients (most with an SM-AHN),17 but they are less frequent in patients with ISM/smoldering SM (SSM).17,18 In advSM, ≥3 or ≥5 mutations were identified in 78% or 41% of patients, respectively. The molecular profile of granulocyte-macrophage colony-forming progenitor cells in KIT D816V+ SM-AHN patients and logical hierarchy analysis showed that somatic mutations in TET2, SRSF2, and ASXL1 precede KIT D816V.19 Inferior survival was observed for SM patients who were grouped based on the presence of combined TET2/DNMT3A/ASXL1 mutations independent of KIT and sole TET2 mutations.20 In fact, KIT D816V and TET2 mutations gave rise to a more aggressive disease in mice compared with that induced by KIT D816V alone.21 In a series of 70 multimutated advSM patients, overall survival (OS) was adversely influenced by the presence and number of mutated genes in the SRSF2/ASXL1/RUNX1 (S/A/R) gene panel.22 In an independent series of 19 advSM patients, non-KIT mutations were frequent (79%), particularly in TET2, ASXL1, and CBL. Presence of ASXL1 and/or CBL mutations or occurrence of ≥3 non-KIT mutations was independently associated with significantly inferior OS, but not leukemia-free survival.23 A recent study found that the presence of ≥1 multilineage mutation in the S/A/R gene panel and/or the EZH2 gene was the sole independent predictor for progression-free survival (PFS) and OS.24
Impact of cytogenetic abnormalities
In a study of SM, cytogenetic abnormalities were identified only in SM-AHN patients (22%).25 Progression to MC leukemia (MCL) or secondary AML and shortened OS were associated with complex karyotype or monosomies and were independent of mutation status. Shah et al26 retrospectively identified an abnormal karyotype in 15% of SM patients, with the highest incidence (26%) in individuals with SM-AHN. Multivariate analysis in SM-AHN patients revealed independent prognostic contributions from adverse mutations, anemia, and thrombocytopenia, but not from abnormal karyotype.
Clinicopathologic pearls in the diagnosis of SM-AHN and MCL variants
SM-AHN
Multilineage involvement of KIT D816V and the presence of additional somatic mutations mirror the clinicopathologic heterogeneity of advSM (Table 1; Figure 1).10,27-29 This is particularly true in SM-AHN, where the extent of MC vs AHN involvement of the marrow or other organs may be similar or discordant in an individual. It can be difficult to determine whether the SM or AHN component is primarily responsible for organ damage and therefore which requires more immediate therapy. In addition, neoplastic MCs are not equal-opportunity organ offenders; a patient with a low BM MC burden and normal blood counts may exhibit extensive liver involvement, resulting in hepatomegaly with liver dysfunction (most commonly elevated alkaline phosphatase [AP]), splenomegaly, and variable portal hypertension and/or ascites.29
Clinicopathologic pearls in the diagnosis of SM variants
| PB MC, % | PB blasts, % | Cytopenias | -cytoses | BM aspirate MCs, % | Dysplasia | BM biopsy cellularity away from MC aggregates | BM biopsy MCs, % | Serum tryptase, ng/mL | Abnormal karyotype | PB KIT D816V qPCR | BM KIT D816V qPCR | Additional somatic mutations* | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ISM | 0 | 0 | No | No | <5 | No | Normo | <20 | ≥20† | No | Low | Low | ± |
| SSM | 0 | 0 | No | No | <5 | Var‡ | Normo/hyper§ | >30 | ≥200 | No | Low | High | ± |
| ASM | 0 | 0 | Yes|| | No | <20¶ | No | Normo | >50 | ≥200 | No | Low | High | + |
| MCL# | Var# | 0 | Var|| | Vara | ≥20 | No | Normo | >50 | ≥200 | No | Var | High | + |
| ISM-AHN | 0 | Varb | Yes|| | Vara | <5 | Var | Hyper | <20-50 | >20-50 | Yes | Highc | High | ++ |
| ASM/MCL-AHN | Var# | Varb | Yes|| | Vara | ≥ 20 | Var | Hyper | >50 | ≥200 | Yes | High | High | +++ |
| PB MC, % | PB blasts, % | Cytopenias | -cytoses | BM aspirate MCs, % | Dysplasia | BM biopsy cellularity away from MC aggregates | BM biopsy MCs, % | Serum tryptase, ng/mL | Abnormal karyotype | PB KIT D816V qPCR | BM KIT D816V qPCR | Additional somatic mutations* | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ISM | 0 | 0 | No | No | <5 | No | Normo | <20 | ≥20† | No | Low | Low | ± |
| SSM | 0 | 0 | No | No | <5 | Var‡ | Normo/hyper§ | >30 | ≥200 | No | Low | High | ± |
| ASM | 0 | 0 | Yes|| | No | <20¶ | No | Normo | >50 | ≥200 | No | Low | High | + |
| MCL# | Var# | 0 | Var|| | Vara | ≥20 | No | Normo | >50 | ≥200 | No | Var | High | + |
| ISM-AHN | 0 | Varb | Yes|| | Vara | <5 | Var | Hyper | <20-50 | >20-50 | Yes | Highc | High | ++ |
| ASM/MCL-AHN | Var# | Varb | Yes|| | Vara | ≥ 20 | Var | Hyper | >50 | ≥200 | Yes | High | High | +++ |
The pathology workup for SM includes examination of PB and BM aspirate smears, BM biopsy, special stains to assess fibrosis (e.g. reticulin, trichrome, Gomori), and immunohistochemistry to quantify and phenotype MCs (tryptase, CD117, CD25, ±CD30) and assess AHN (eg, CD34, CD3, CD14, CD20, CD138, CD61, determined by histopathologic assessment). Because the MC infiltrate in ISM-AHN can be subtle, a tryptase immunohistochemical stain should be performed in select myeloid neoplasms such as AML with t(8;21) and MDS/MPNs such as CMML. As a corollary, in a series of >1500 patients with a WHO-based diagnosis of CMML, NGS identified KIT D816V in 6% of cases (T. Haferlach, Munich Leukemia Laboratory, Munich, Germany, personal communication), although KIT D816V positivity is not always accompanied by SM. Fibrosis is present in association with MC aggregates; fibrosis outside of MC aggregates suggests an AHN. Ancillary testing such as flow cytometry (preferred for assessment of CD2 on MC and phenotypic characterization of AHNs), karyotype, PDGFRA fluorescence in situ hybridization if eosinophilia, KIT D816V quantitative PCR (qPCR), and NGS myeloid gene panels are further discussed in National Comprehensive Cancer Network guidelines.61
Hyper, hypercellular; normo, normocellular; var, variable.
Additional somatic mutations commonly include SRSF2, ASXL1, RUNX1, CBL, JAK2, and EZH2. Only ∼25% of ISMs and SSMs have an additional somatic mutation present (±). In ASM and MCL, approximately one-half will have additional mutations (+), whereas in SM-AHN, additional mutations are common (++ or +++ depending on number of mutations).
Serum tryptase level is mildly increased in patients with ISM, but may be normal when there is low-level BM involvement.
Some dysplasias (or signs of myeloproliferation) may be seen in non-MC lineages, but criteria for diagnosis of an AHN are not met. Blood counts are normal or only slightly abnormal.
Hypercellularity in the BM away from MC aggregates may be seen.
Cytopenias are commonly seen in ASM and acute MCL, as C findings (hemoglobin <10 g/dL, absolute neutrophil count <1.0 × 109/L, platelets <100 × 109/L), but can also be seen secondary to an AHN. Chronic MCL, by definition, lacks C findings.
ASM in transformation denotes 5%-19% MCs on BM aspirate smear, which has a high rate of progression to MCL.
Most MCLs are aleukemic, as defined by <10% circulating MCs in PB; ≥10% circulating MCs in PB is leukemic MCL. Other subtypes include de novo (primary) or secondary MCL, acute or chronic MCL, and MCL with or without an AHN.
A mild increase in eosinophils (<1.5 × 109/L) may be seen in MCL. Other cytoses, including eosinophilia ≥1.5 × 109/L, suggest an AHN. Absolute monocytes >1.0 × 109/L and >10% monocytes on PB differential raise concern for CMML. Either an absolute monocytosis or >10% monocytes, but not both, can suggest MDS/MPN unclassifiable.
Depending on the type of AHN, there may or may not be circulating blasts.
Discordantly high KIT D816V VAF in the PB when there is a low MC burden in the BM is a surrogate for multilineage involvement/AHN.
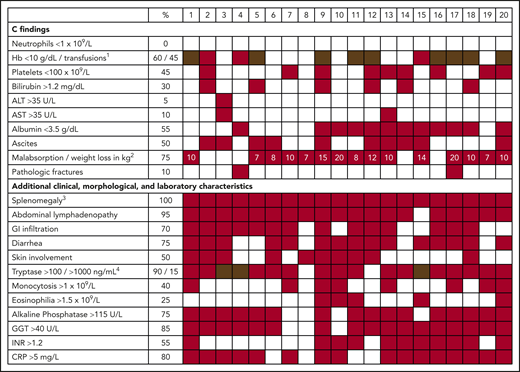
Clinical heterogeneity of patients with advSM. Clinical, morphological, and laboratory characteristics (including C findings) of 20 patients with advSM. 1Anemia (red), transfusion-dependent anemia (brown). 2Numbers in red boxes reflect weight loss in kilograms. 3Splenomegaly includes patients in which splenomegaly would qualify as a B finding and patients with hypersplenism (eg, thrombocytopenia), which would qualify as a C finding. A clear distinction can be challenging in individual patients. 4Tryptase >100 ng/mL (red), >1000 ng/mL (brown). ALT, alanine aminotransferase; AST, aspartate aminotransferase; CRP, C-reactive protein; GGT, gamma-glutamyl transferase; GI, gastrointestinal; Hb, hemoglobin; INR, international normalized ratio.
Clinical heterogeneity of patients with advSM. Clinical, morphological, and laboratory characteristics (including C findings) of 20 patients with advSM. 1Anemia (red), transfusion-dependent anemia (brown). 2Numbers in red boxes reflect weight loss in kilograms. 3Splenomegaly includes patients in which splenomegaly would qualify as a B finding and patients with hypersplenism (eg, thrombocytopenia), which would qualify as a C finding. A clear distinction can be challenging in individual patients. 4Tryptase >100 ng/mL (red), >1000 ng/mL (brown). ALT, alanine aminotransferase; AST, aspartate aminotransferase; CRP, C-reactive protein; GGT, gamma-glutamyl transferase; GI, gastrointestinal; Hb, hemoglobin; INR, international normalized ratio.
Quantitative assessment of BM MC infiltration and serum tryptase level30 are relevant markers for SM diagnosis but do not always correspond with SM subtype or disease burden. In addition, mild increases in the basal serum tryptase level can be observed in other myeloid neoplasms, hereditary α-tryptasemia, and MC activation syndrome, where this level can also be used as a dynamic marker of an acute flare of mediator symptoms or anaphylaxis.30-32
Diagnosis of ISM (low MC burden and tryptase level) and MCL (high MC burden and tryptase level) is relatively straightforward (Table 1). However, SM-AHN may present with a low MC burden and low serum tryptase level, but a high AHN disease burden (eg, marked monocytosis in blood and BM), which is best quantified by a discordantly high KIT D816V VAF of up to 20% to 50%. Still, one of the most reliable indicators of progressive SM is a steadily increasing basal serum tryptase level. Clinical and morphologic indicators of a concomitant AHN include -cytoses, cytopenias, dysplasia, elevated lactate dehydrogenase, splenomegaly, hypercellular BM, and osteosclerosis; this should prompt NGS testing to further characterize the AHN. SM may be overlooked by pathologists in some patients with myeloid neoplasms, particularly CMML, CEL, and AML, because of inadequate staining of core biopsy sections.33,34 In our experience, detection of KIT D816V in the PB at a VAF >2% to 5% suggests multilineage involvement/AHN in ISM or masked SM in myeloid neoplasms.
MCL
Recently, it was found that the leukemia-initiating stem cell in MCL resides within the CD34+/CD38− fraction, but not in CD34+/CD38+ progenitors or bulk KIT+/CD34− MCs.35 MCL is a histopathologic diagnosis based on the presence of ≥20% MCs on a BM aspirate smear (not BM core biopsy).9 The diagnosis of MCL does not depend on the presence of organ damage (C findings), but >90% of MCL patients ultimately demonstrate this. Traditionally, MCL has been divided into leukemic and aleukemic variants, with <10% and ≥10% MCs in PB, respectively; however, the leukemic variant is rare.9,36 The heterogeneous clinical presentation and disease course suggest alternative distinctions such as the following:
MCL with or without AHN
In addition to morphologically clearly visible signs of proliferation and/or dysplasia, rapid and robust results are provided by a high PB KIT D816V VAF and presence of additional somatic mutations without circulating MCs.
Acute and chronic MCL
These are defined by presence or absence of C findings, respectively.37 The distinction may overlap in a substantial proportion of patients with MCL with presence or absence of an AHN and/or additional somatic mutations. In addition, some cases of chronic MCL present with well-differentiated morphologic and immunophenotypic features together with imatinib-sensitive germ line or somatic KIT mutations (exons 8-11).37,38
De novo (primary) and secondary MCL
These represent either immediate diagnosis of MCL (primary) or progression from another subtype of SM to secondary MCL with or without an AHN.
Jawhar et al39 reported on the clinical and molecular characteristics of 28 MCL patients (MCL-AHN; n = 20; 71%). MCL was de novo or secondary in ∼50% each. Median BM core biopsy MC infiltration was 65%, and median serum tryptase level was 520 ng/mL. C findings were identified in >90% of patients. Mutations in KIT (D816V, n = 19; D816H/Y, n = 5; F522C, n = 1) were detected in 25 (89%) of 28 patients. S/A/R positivity (52%) adversely affected response to treatment, progression to secondary MCL or AML during treatment, and, in a multivariate analysis, OS. Median OS from MCL diagnosis was 17 months, compared with 44 months, in 124 patients with advSM other than MCL.
New molecularly-anchored prognostic scoring systems
Since consensus criteria for the diagnosis and classification of mastocytosis were developed in 2001,40,41 molecular analyses have enhanced the understanding of differences between SM variants. From a genetics perspective, 3 major subgroups of SM have been defined: (1) KIT D816V restricted to the MC lineage, as found in a majority of patients with ISM and frequently nonprogressive SM and only rarely in those with more advanced forms of SM (potentially representing slowly progressive advSM [eg, chronic MCL35 ]); (2) multilineage involvement of KIT as the basis of progressive ISM, SSM, or advSM; and (3) multilineage involvement of KIT D816V plus additional somatic mutations as the basis of ASM/MCL with or without an AHN (Figure 2). Recently, single-cell analysis identified the KIT D816V mutation in early hematopoietic stem (HSC) and progenitor cells in addition to MCs, suggesting that KIT D816V may not be restricted to the MC lineage.42
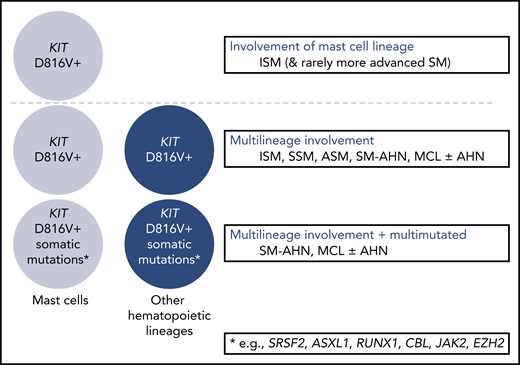
Genetic complexity among systemic mastocytosis subtypes. Clinical characteristics, morphology, and genetics should be seen as complementary tools for subtyping of SM. The presence of multilineage KIT and/or a multimutated molecular profile may contribute to a more advanced presentation of SM and/or disease evolution. For example, multilineage ISM is prone to progression to advanced SM, and prognosis may be equal or even worse than in MCL without multilineage involvement, additional somatic mutations, or C findings.
Genetic complexity among systemic mastocytosis subtypes. Clinical characteristics, morphology, and genetics should be seen as complementary tools for subtyping of SM. The presence of multilineage KIT and/or a multimutated molecular profile may contribute to a more advanced presentation of SM and/or disease evolution. For example, multilineage ISM is prone to progression to advanced SM, and prognosis may be equal or even worse than in MCL without multilineage involvement, additional somatic mutations, or C findings.
The combination of clinical characteristics (AP, spleen volume) and somatic mutations beyond KIT D816V formed the basis of the first SM prognostic risk scoring system.43 Among patients with ISM, REMA found that serum β2-microglobulin levels >2.5 µg/mL, BM KIT D816V VAF ≥1%, and mutations in ASXL1, RUNXL1, and/or DNMT3A (A/R/D) with VAFs ≥30% were the best combination of independent predictors for PFS; mutation of A/R/D genes was the only independent predictor for OS.18
A multivariate analysis of clinical variables derived from 580 SM patients identified age >60 years, advSM, platelet count <150 × 109/L, anemia below sex-adjusted normal, and increased AP as independent risk factors for survival in a clinical risk model.44 Adverse mutations (ASXL1, RUNX1, and NRAS), advSM, thrombocytopenia, increased AP, and age >60 years were identified as independent prognostic variables in a hybrid clinical-molecular risk model (Mayo Alliance Prognostic System; MAPS). An updated analysis of the same cohort without the World Health Organization (WHO) classification or genetic information was used to generate a model consisting of 5 risk groups (WHO class-independent Mayo Alliance Prognostic System).45 The prognostic information from adverse mutations was limited, because they almost exclusively clustered with very high-risk and high-risk disease.
The Mutation-Adjusted Risk Score (MARS) was derived from a clinical and molecular evaluation of 383 patients with advSM.46 Age >60 years, hemoglobin <10 g/dL, platelets <100 × 109/L, presence of 1 high molecular risk mutation in the S/A/R gene panel, and ≥2 high molecular risk gene mutations were independent risk factors for OS. Three risk categories were defined: low (median OS not reached), intermediate (median OS, 3.9 years), and high (median OS, 1.9 years). The mutation-adjusted risk score was also predictive for leukemia-free survival.46
The International Prognostic Scoring System for Mastocytosis (IPSM) comprises the largest cohort of SM patients (N = 1639), taken from the European Competence Network on Mastocytosis (ECNM) multicenter registry.47 The IPSM confirmed the prognostic value of the WHO classification and identified independent prognostic factors for patients with non-advSM (age ≥60 years, AP ≥100 U/L) and advSM (age ≥60 years, tryptase level ≥125 ng/mL, leukocyte count ≥16 × 109/L, hemoglobin <11 g/dL, platelet count ≤100 × 109/L, and lack of skin involvement).47 This permitted further stratification of non-advSM and advSM into subgroups with significant differences in PFS and OS.
REMA is validating a new global prognostic scoring system (Alberto Orfao, Universidad de Salamanca, personal communication, December 2019) and comparing its ability to discriminate PFS and OS vs the aforementioned scoring systems (summarized in Table 2). These prognostic scoring systems incorporate laboratory results obtained in routine practice and, with the increasing availability of NGS panels, should provide more precision in the risk assessment of SM progression to help guide the timing and intensity of treatment.
Prognostic scoring systems in SM
| Parameter | REMA (for ISM) | MAPS | IPSM | MARS | GPS | |||
|---|---|---|---|---|---|---|---|---|
| OS | PFS | Nonadvanced | Advanced | OS | PFS | |||
| WHO (advSM) | + | |||||||
| Age ≥60 y | + | + | + | + | ||||
| Anemia, g/dL | ||||||||
| ≤10 | + | |||||||
| ≤11 | + | + | ||||||
| Thrombocytopenia, × 109/L | ||||||||
| <100 | + | + | + | |||||
| <150 | + | |||||||
| Leukocytosis, × 109/L | ||||||||
| ≥16 | + | |||||||
| Increased serum markers | ||||||||
| Baseline serum tryptase | + | + | ||||||
| β2-microglobulin | + | + | ||||||
| Alkaline phosphatase | + | + | + | |||||
| Genetics | ||||||||
| BM KIT D816V VAF ≥1% | + | |||||||
| Additional somatic mutations | + (A/R/D) | + (A/R/D) | + (A/R/NRAS) | + (S/A/R) | + (S/A/R/D) | |||
| Parameter | REMA (for ISM) | MAPS | IPSM | MARS | GPS | |||
|---|---|---|---|---|---|---|---|---|
| OS | PFS | Nonadvanced | Advanced | OS | PFS | |||
| WHO (advSM) | + | |||||||
| Age ≥60 y | + | + | + | + | ||||
| Anemia, g/dL | ||||||||
| ≤10 | + | |||||||
| ≤11 | + | + | ||||||
| Thrombocytopenia, × 109/L | ||||||||
| <100 | + | + | + | |||||
| <150 | + | |||||||
| Leukocytosis, × 109/L | ||||||||
| ≥16 | + | |||||||
| Increased serum markers | ||||||||
| Baseline serum tryptase | + | + | ||||||
| β2-microglobulin | + | + | ||||||
| Alkaline phosphatase | + | + | + | |||||
| Genetics | ||||||||
| BM KIT D816V VAF ≥1% | + | |||||||
| Additional somatic mutations | + (A/R/D) | + (A/R/D) | + (A/R/NRAS) | + (S/A/R) | + (S/A/R/D) | |||
Adapted from Muñoz-González and Orfao85 with permission.
A, ASXL1; D, DNMT3A; GPS, global prognostic scoring system; R, RUNX1; S, SRSF2.
Treatment
KIT D816V, a primary oncogenic driver of MC differentiation, proliferation, and survival, is an attractive target because of its high frequency in SM.9,48,49 It confers primary resistance against the tyrosine kinase inhibitors imatinib and masitinib.50,51 Despite their low 50% inhibitory concentration values against KIT D816V, nilotinib and dasatinib lack significant clinical activity.52,53 Imatinib is US Food and Drug Administration–approved for ASM patients negative for KIT D816V or with unknown KIT mutation status; however, this is relevant to few advSM patients. Imatinib-sensitive KIT mutations in the extracellular (eg, deletion of codon 419 on exon 8 or p.A502_Y503dup in exon 9), transmembrane (eg, F522C), or juxtamembrane (eg, V560G) domain are found in <1% of all advSM cases, but seem to be enriched in cases of well-differentiated SM.38,54
KIT inhibition: results from the midostaurin and avapritinib trials
Table 3 lists key efficacy and safety data from the pivotal phase 2 D2201 midostaurin registration study55 that led to its regulatory approval in 2017, with data from a smaller investigator-initiated trial,56 and the most recent available results from the ongoing phase 1 dose-escalation and -expansion study of avapritinib in advSM.57 The switch-control inhibitor ripretinib (DCC-2618)58 is currently undergoing phase 1 evaluation for patients with advSM and SSM. It is important to highlight that the efficacy of midostaurin and avapritinib has been adjudicated using different response criteria (Table 3).55-57,59,60 With this caveat, the ongoing phase 1 study of avapritinib has revealed an overall response rate of 77%, including a CR plus CRh rate of 23%, associated with marked reduction of measures of MC burden (eg, percentage of BM MCs, tryptase level, and KIT D816V VAF [which can reflect both the MC and AHN components]).57 National Comprehensive Cancer Network guidelines are now available to guide treatment approaches to SM, including the use of midostaurin and enrollment in clinical trials using KIT inhibitors or other agents.61
Summary of midostaurin and avapritinib efficacy and safety outcomes
| Midostaurin55 | Avapritinib57 | |
|---|---|---|
| Trial design | Phase 2, single arm, open label | Phase 1, dose escalation and expansion |
| Patients, n | 116 | Dose escalation, 37 |
| Dose extension, 32 | ||
| Evaluable for response, n | 89 | 39 |
| Response criteria | Modified Valent and modified Cheson* | Modified IWG-MRT-ECNM† |
| ORR, % | 60 (MR + PR) | 77 (CR + CRh + PR + CI) |
| Response subcategory, % | MR 45 | CR 8 |
| CR 0 | CRh 15 | |
| Incomplete remission 21 | PR 46 | |
| Pure clinical response 17 | CI 8 | |
| Unspecified 7 | SD 23 | |
| PR 15 | PD 0 | |
| SD 12 | ||
| PD 11 | ||
| Not evaluable 17 | ||
| Note: 26% of patients previously treated with midostaurin | ||
| Post hoc exploratory efficacy analysis by IWG-MRT-ECNM criteria using algorithmic approach, %† | ||
| FDA (CR + PR) | 17 (CR [2] + PR [15]) | Not applicable |
| EMA (CR + PR + CI) | 28 (CR [1] + PR [15] + CI [12]) | |
| Response rate by advSM subgroup, % | ||
| ASM | 75 | 100 |
| SM-AHN | 58 | 75 |
| MCL | 50 | 75 |
| Patients with ≥50% decrease in BM MCs, % | 57 | 93 |
| Patients with ≥50% decrease in serum tryptase, % | 60 | 100 |
| Evaluable patients with ≥35% decrease in spleen volume, % | 26 | 81 |
| AE profile (any grade/grade 3-4), % | Nausea 79/6 | Periorbital edema 75/4 |
| Vomiting 66/6 | Diarrhea 41/1 | |
| Diarrhea 54/8 | Nausea 38/4 | |
| Peripheral edema 34/4 | Fatigue 36/7 | |
| Abdominal pain 28/3 | Peripheral edema 33/0 | |
| Fatigue 28/9 | Vomiting 32/4 | |
| Pyrexia 27/6 | Cognitive effects 32/4 | |
| Constipation 24/1 | Hair color changes 29/1 | |
| Headache 23/2 | Arthralgia 20/1 | |
| Neutropenia 48/24 | Neutropenia 12/10 | |
| Anemia 63/41 | Anemia 55/29 | |
| Thrombocytopenia 52/29 | Thrombocytopenia 35/23 | |
| Intracranial bleeding (ICB) in 7 patients; 5 of 7 resumed therapy; 1 ICB in setting of severe head trauma; dose modifications for thrombocytopenia instituted to mitigate ICB |
| Midostaurin55 | Avapritinib57 | |
|---|---|---|
| Trial design | Phase 2, single arm, open label | Phase 1, dose escalation and expansion |
| Patients, n | 116 | Dose escalation, 37 |
| Dose extension, 32 | ||
| Evaluable for response, n | 89 | 39 |
| Response criteria | Modified Valent and modified Cheson* | Modified IWG-MRT-ECNM† |
| ORR, % | 60 (MR + PR) | 77 (CR + CRh + PR + CI) |
| Response subcategory, % | MR 45 | CR 8 |
| CR 0 | CRh 15 | |
| Incomplete remission 21 | PR 46 | |
| Pure clinical response 17 | CI 8 | |
| Unspecified 7 | SD 23 | |
| PR 15 | PD 0 | |
| SD 12 | ||
| PD 11 | ||
| Not evaluable 17 | ||
| Note: 26% of patients previously treated with midostaurin | ||
| Post hoc exploratory efficacy analysis by IWG-MRT-ECNM criteria using algorithmic approach, %† | ||
| FDA (CR + PR) | 17 (CR [2] + PR [15]) | Not applicable |
| EMA (CR + PR + CI) | 28 (CR [1] + PR [15] + CI [12]) | |
| Response rate by advSM subgroup, % | ||
| ASM | 75 | 100 |
| SM-AHN | 58 | 75 |
| MCL | 50 | 75 |
| Patients with ≥50% decrease in BM MCs, % | 57 | 93 |
| Patients with ≥50% decrease in serum tryptase, % | 60 | 100 |
| Evaluable patients with ≥35% decrease in spleen volume, % | 26 | 81 |
| AE profile (any grade/grade 3-4), % | Nausea 79/6 | Periorbital edema 75/4 |
| Vomiting 66/6 | Diarrhea 41/1 | |
| Diarrhea 54/8 | Nausea 38/4 | |
| Peripheral edema 34/4 | Fatigue 36/7 | |
| Abdominal pain 28/3 | Peripheral edema 33/0 | |
| Fatigue 28/9 | Vomiting 32/4 | |
| Pyrexia 27/6 | Cognitive effects 32/4 | |
| Constipation 24/1 | Hair color changes 29/1 | |
| Headache 23/2 | Arthralgia 20/1 | |
| Neutropenia 48/24 | Neutropenia 12/10 | |
| Anemia 63/41 | Anemia 55/29 | |
| Thrombocytopenia 52/29 | Thrombocytopenia 35/23 | |
| Intracranial bleeding (ICB) in 7 patients; 5 of 7 resumed therapy; 1 ICB in setting of severe head trauma; dose modifications for thrombocytopenia instituted to mitigate ICB |
CI, clinical improvement; CR, complete response; CRh, CR with partial hematologic recovery; EMA, European Medicines Agency; FDA, US Food and Drug Administration; IWG, International Working Group; MR, major response; MRT, Myeloproliferative Neoplasms Research and Treatment; ORR, overall response rate; PD, progressive disease; PR, partial response; SD, stable disease.
Responses need to be confirmed for 8 wk.
Responses need to be confirmed for 12 wk.
Outstanding questions related to SM-AHN in KIT inhibitor trials
In the studies of midostaurin and avapritinib, a majority of advSM patients (∼70%) were SM-AHN by central pathology review.55-57 In some cases, the AHN component was missed by the local site, illustrating the potential for underdiagnosis of this advSM variant in real-world practice (Table 1).
Response rates in the KIT inhibitor studies are anchored to SM end points such as reversion of organ damage (centrally adjudicated as related to MC disease), tryptase level, and BM MC burden.55-57 Although a comprehensive assessment of the clinicopathologic response of the AHN component to KIT inhibition has not yet been undertaken, some general observations can be made: the response of SM and AHN can be highly variable among patients, between SM and AHN in the same patient, and even among various AHN lineages (eg, eosinophils and monocytes). In most advSM patients with eosinophilia (eg, SM-CEL), KIT inhibition results in rapid and complete normalization of PB and BM eosinophilia.55,56 However, in those with SM-CMML, for example, there are fewer reductions in PB and BM monocytes.55,56 The biologic basis for the sensitivity of eosinophils and relative insensitivity of monocytes to KIT inhibitors is not well understood.
It is currently unknown whether KIT inhibition affects the natural history of advSM patients. In the Mayo series of SM, median OS of SM-AHN patients was 24 months and varied by AHN subtype (SM-MPN, 31 months; SM-CMML, 15 months; SM-MDS, 13 months).28,62 In the midostaurin trial,55 median OS was 28.7 months in all advSM patients and 20.7 months in the subgroup of SM-AHN patients. In the ongoing avapritinib trial,57 which has a shorter follow-up, median OS of individuals with SM-AHN has not yet been reached. The Kaplan-Meier estimate of 2-year OS for SM-AHN patients was 49% and 70%, respectively, for SM-AHN patients enrolled in the midostaurin and avapritinib trials. Comparisons of OS between interventional trials and retrospective case series are confounded by several factors. First, OS in the trials was measured from the time of treatment initiation, not from the time of diagnosis, as in the Mayo series. Second, patients were not matched for baseline host- and disease-related factors such as patient age and comorbidities, type and stage of AHN, comutation status, prior therapy, and measures of SM burden. Interestingly, the rate of progression to secondary AML in the midostaurin trial was 11%,55 the same rate as in the collective cohort of advSM patients in the Mayo series.62 In all studies, progression to AML was highly enriched in patients with SM-AHN compared with patients with ASM, consistent with the observation that the AHN (rather than SM) component usually drives prognosis. To date, no data have established that KIT inhibition can alter the rate of progression to AML or extend OS in patients with advSM, including those with SM-AHN. However, ongoing durable responses and survival of >3 to 5 years in some MCL patients treated with midostaurin (with similar responses emerging in avapritinib-treated MCL patients) suggest that a survival signal may be emerging in this poorest-risk group of patients whose OS is historically <6 to 18 months (but can be longer in patients without S/A/R mutations or an AHN).39
Lessons learned from the KIT inhibitor trials
Trial experiences with midostaurin and avapritinib are prompting a reevaluation of how to optimize the IWG-MRT-ECNM response criteria for advSM.60 First, KIT inhibition can produce myelosuppression, particularly in patients with preexisting cytopenias.55-57 Some patients in the phase 1 avapritinib trial met all criteria for a CR (including disappearance of BM MC aggregates) with the exception of cytopenias attributed to the drug.57 IWG-MRT-ECNM criteria were modified for the avapritinib trial to include the aforementioned CRh category. CRh is similar to CR but is defined by ≥1 cytopenias (absolute neutrophil count ≥0.5 × 109/L, Hb ≥8 g/dL, and platelet count ≥50 × 109/L) that are unrelated to SM (eg, KIT inhibition–related myelosuppression, persistence of the AHN, or both). Long-term follow-up will help determine whether achievement of a CR vs CRh will translate into different outcomes, as has been observed in AML patients receiving cytarabine-based therapy whose relapse-free survival was superior in those achieving a CR vs a CR with incomplete platelet recovery.63
Both midostaurin and avapritinib treatment have resulted in reproducible histopathologic changes in the BM, reflecting the effects of KIT inhibition (Figure 3). In responding patients, dense MC aggregates decrease in number and frequently assume an interstitial pattern, with fewer MCs over the course of 3 to 11 months (or longer). Concurrently, the marrow becomes more hypocellular, reflecting drug-related myelosuppression, and may subsequently normalize cellularity. Atypical MCs typically revert to a normal morphology and lose expression of CD25, which often occurs in parallel with a decrease in serum tryptase level. These dynamic BM changes need to be recognized by pathologists who will be tasked with interpreting the treatment effects of KIT inhibitors in SM patients.
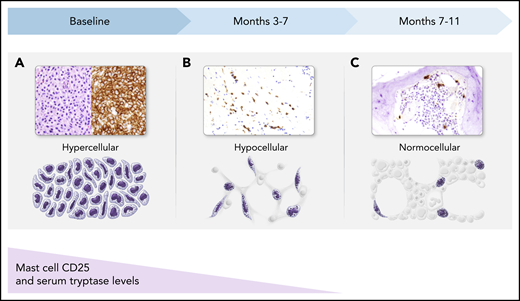
BM response to KIT inhibition in patients with SM. (A) At baseline, atypical (hypogranular, immature, and spindle-shaped) MCs in dense aggregates fill the BM (hematoxylin and eosin stain and CD25 immunohistochemical stain). (B) After a few months of therapy, dense aggregates are few in number, and predominantly loose clusters of MCs are present, with fewer atypical MCs (CD117 immunohistochemical stain). (C) By several months of therapy, only interstitial scattered single MCs remain, which are mostly small, round, and well granulated, with few atypical spindle-shaped MCs (tryptase immunohistochemical stain). During this same time period, MCs that express CD25 initially will lose expression of this aberrant marker, reverting to a normal MC phenotype. Serum tryptase levels similarly decline and generally correlate with MC burden in the BM. All magnification ×40.
BM response to KIT inhibition in patients with SM. (A) At baseline, atypical (hypogranular, immature, and spindle-shaped) MCs in dense aggregates fill the BM (hematoxylin and eosin stain and CD25 immunohistochemical stain). (B) After a few months of therapy, dense aggregates are few in number, and predominantly loose clusters of MCs are present, with fewer atypical MCs (CD117 immunohistochemical stain). (C) By several months of therapy, only interstitial scattered single MCs remain, which are mostly small, round, and well granulated, with few atypical spindle-shaped MCs (tryptase immunohistochemical stain). During this same time period, MCs that express CD25 initially will lose expression of this aberrant marker, reverting to a normal MC phenotype. Serum tryptase levels similarly decline and generally correlate with MC burden in the BM. All magnification ×40.
Integration of molecular analysis in trials of KIT inhibitors
In the midostaurin and avapritinib trials, the focus on dynamic changes in KIT D816V VAF and serial profiling of myeloid mutations has been informative. Among a cohort of 38 advSM patients treated with midostaurin, Jawhar et al8 found that overall response rate and OS were significantly higher in patients with ≥25% reduction in KIT D816V RNA-expressed allele burden (ie, KIT responders) and in patients without S/A/R mutations. In a multivariate analysis, KIT responder status was the strongest and only on-treatment variable associated with prolonged OS. In the latest update of the avapritinib trial, 88% of patients achieved a >50% reduction in marrow KIT D816V VAF, and 33% exhibited a complete molecular remission of KIT D816V using digital droplet PCR (detection threshold, 0.17%).57 The high rate of molecular remissions is in keeping with the 10-fold greater in vitro potency of avapritinib against KIT D816V compared with midostaurin.64 Although not included in the current IWG-MRT-ECNM response criteria,60 integration of molecular remission and minimal residual disease end points will help determine whether achieving KIT D816V− MRD affects PFS or OS.
Jawhar et al8 reported that progression of midostaurin-treated advSM patients to MCL or secondary AML was associated with ≥1 S/A/R mutations, an increase in the VAF of preexisting mutations, or on-treatment development of new mutations in genes such as K/NRAS, RUNX1, IDH2, and NPM1. In vitro modeling of KIT D816V–transformed Ba/F3 cells treated with midostaurin and avapritinib found that secondary KIT V654A, N655K, and D677N mutations conferred resistance to midostaurin, and the T670I gatekeeper mutation preferentially conferred resistance to avapritinib.65 To date, neither these nor other KIT mutations, except for D816V, have been identified in patients. This parallels the experience of myelofibrosis patients with disease persistence or progression to AML in whom no additional JAK2 mutations besides V617F have been identified.66 Both in vitro data (using single cell–derived myeloid progenitor cells from patients with KIT D816V+ advSM)67 and the phase 1 trial experience57 have demonstrated the activity of avapritinib in patients progressing on midostaurin (including those with S/A/R mutations).
Determination of KIT D816V status by sensitive PCR and extended molecular profiling of KIT inhibitor–treated patients with NGS panels is encouraged before and during KIT inhibitor treatment. This is especially applicable at the time of CR or disease progression. In the case of progression to AML, opportunities for alternative targeted therapies may arise (eg, IDH1, IDH2, BCL-2, or FLT3 inhibitors). Little is known about the changes in clonal architecture of the disease under the selection pressure of KIT inhibition. Single cell–targeted DNA sequencing and transcriptome analysis of bulk marrow or flow-sorted HSCs, MCs, and non-MC lineages will provide critical insight into the cooccurrence (and relative frequency) of mutations in MCs with KIT D816V and in AHN cells with or without KIT D816V. As was recently demonstrated in AML,68,69 these data may help customize combinatorial targeted therapeutic approaches for resistant disease and frontline treatment approaches for advSM patients.
Future study designs
The midostaurin and avapritinib studies provide a foundation for future trial designs and priorities. For advSM patients without an AHN who exhibit refractory/relapsed SM, the addition of other active agents in SM should be considered. In a retrospective French long-term study that included 32 advSM patients, cladribine demonstrated an overall response rate of 50%, corroborating smaller phase 2 studies.70 Cladribine can exhibit rapid debulking activity, but high-grade myelosuppression and opportunistic infections are common. For patients with refractory/relapsed advSM, strategies that combine cladribine with KIT inhibitors may require syncopated treatment schedules, dose reduction of 1 or both agents, or extended cycle lengths to permit hematopoietic recovery. Evaluation of the relevance of novel agents with mechanisms of action not overlapping those of KIT inhibitors, such as antibodies against surface antigens (eg, CD25, CD30, CD33, CD52, CD123, siglec-8) and inhibitors of intracellular signaling pathways (eg, JAK-STAT, PI-3-kinase, BCL-2), to neoplastic MC expansion remains a clinical imperative.71-78
Patients with SM-AHN may benefit from adaptive trial designs, such as that of the BEAT AML Master Clinical Trial,79 which recognizes numerous AML subtypes based on molecular heterogeneity, instead of 1 disease, and accordingly randomizes patients to specific treatment cohorts based on NGS screening results. This approach is translatable to SM-AHN, where screening of the type and stage of AHN as well NGS testing would inform the selection of agents in combination with a KIT inhibitor. Figure 4 highlights several variations of this theme that could be considered in the design of such a basket trial for various SM-AHNs.
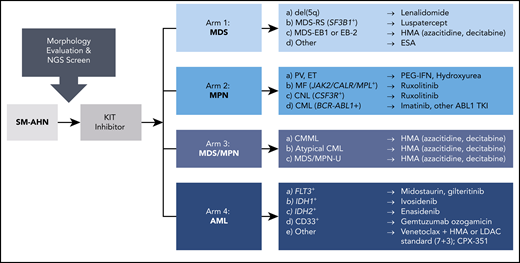
A precision medicine, adaptive trial scheme for diverse SM-AHN variants. Baseline morphologic evaluation, serum tryptase level, KIT D816V VAF, and NGS are used to characterize the burden of SM as well as the type and stage of AHN (if present). This information is also used to stratify patients into potential treatment arms with therapies based on the identification of druggable targets. CML, chronic myeloid leukemia; CNL, chronic neutrophilic leukemia; ESA, erythropoiesis-stimulating agent; ET, essential thrombocythemia; HMA, hypomethylating agent; LDAC, low-dose cytarabine; MDS EB-1 or EB-2, MDS with excess blasts-1 or excess blasts-2; MDS-RS, MDS with ring sideroblasts; MF, myelofibrosis; MPN-U, MPN unclassifiable; PEG-IFN, pegylated interferon; PV, polycythemia vera; TKI, tyrosine kinase inhibitor.
A precision medicine, adaptive trial scheme for diverse SM-AHN variants. Baseline morphologic evaluation, serum tryptase level, KIT D816V VAF, and NGS are used to characterize the burden of SM as well as the type and stage of AHN (if present). This information is also used to stratify patients into potential treatment arms with therapies based on the identification of druggable targets. CML, chronic myeloid leukemia; CNL, chronic neutrophilic leukemia; ESA, erythropoiesis-stimulating agent; ET, essential thrombocythemia; HMA, hypomethylating agent; LDAC, low-dose cytarabine; MDS EB-1 or EB-2, MDS with excess blasts-1 or excess blasts-2; MDS-RS, MDS with ring sideroblasts; MF, myelofibrosis; MPN-U, MPN unclassifiable; PEG-IFN, pegylated interferon; PV, polycythemia vera; TKI, tyrosine kinase inhibitor.
What is the role of allogeneic HSCT in the age of KIT inhibitors?
The encouraging efficacy of KIT inhibitors may influence decision making about allogeneic HSC transplantation (HSCT). The largest retrospective series consists of 57 patients (SM-AHN, n = 38; ASM, n = 7; MCL, n = 12).80 Three-year OS was 74% for patients with SM-AHN, 43% for those with ASM, and 17% for those with MCL. Adverse prognostic factors for OS were diagnosis of MCL and the use of reduced-intensity vs myeloablative conditioning; the latter is troublesome, because advanced age prevents myeloablative allografting in many patients. Although not matched for baseline characteristics, 3-year OS for midostaurin-treated patients was relatively better for ASM (65%) and MCL patients (26%), but lower for SM-AHN patients (44%), compared with the transplantation experience.55 The latest update of the phase 1 avapritinib study shows 2-year OS survival rates of 70%, 100%, and 88% for SM-AHN, ASM, and MCL patients, respectively.57 These data suggest that among advSM variants, SM-AHN patients may be the preferred group for consideration of allogeneic HSCT, but type of AHN and disease status are important factors in the final decision.
In patients with a suitable donor, we generally favor undertaking HSCT at the time of best response after KIT inhibition (with or without AHN-directed therapy).81 However, no prospective data are available to guide the optimal cytoreductive approach or timing of HSCT. Given the potential for KIT inhibitors to induce CR of the SM component, the potential for discordant progression of the AHN, including clonal evolution,8 which may herald relapse and preclude an opportunity for HSCT, should not be overlooked.
Nonmyeloablative conditioning strategies, including anti-CD117 antibodies, are currently being evaluated. For example, an anti-CD117 antibody (AMG 191), either naked or conjugated to saporin, depleted normal (and/or MDS) HSCs, permitting engraftment of normal donor human HSCs in a xenograft mouse model.82,83 If active against neoplastic MCs, these antibodies could therefore serve a dual purpose in advSM, both at time of transplantation as well as for prevention of relapse.81
Conclusion
Advances in the genetic profiling of KIT D816V and myeloid-associated gene mutations have defined the concepts of multilineage KIT involvement and multimutated disease, permitting a more granular explanation for SM disease heterogeneity than that allowed by the WHO classification alone. Similarly, novel hybrid prognostic scoring systems that combine clinical and molecular variables have generated more accurate stratification of disease outcomes, which should facilitate treatment decision making. Although KIT inhibition is now validated as a therapeutic paradigm for advSM, SM-AHN remains a formidable challenge, with numerous unresolved questions related to diagnosis and management, especially the role of HSCT. Collaboration between biopharma and ECNM,84 the American Initiative on Mast Cell Diseases, and patient advocacy groups will be required to address the unmet research needs of this rare disease population.
Acknowledgments
The authors thank the members of ECNM and the American Initiative on Mast Cell Diseases for their collaborative efforts, including Javier Muñoz-González and Alberto Orfao for providing Table 2 on prognostic scoring systems. The authors also thank Evelyn Lockhart and Juliana Schwaab for their illustrations.
J.G. is supported by the Charles and Ann Johnson Foundation and Stanford Cancer Institute Clinical Innovation Fund; T.I.G. is supported by the ARUP Institute for Clinical and Experimental Pathology; and A.R. is supported by the Deutsche José Carreras Leukämie-Stiftung.
Authorship
Contribution: A.R., T.I.G., and J.G. contributed to the writing of the manuscript and creation of figures and tables.
Conflict-of-interest disclosure: J.G. has served on advisory boards and chairman of study steering committees for and received honoraria and funding for the conduct of clinical trials from Novartis, Blueprint Medicines, and Deciphera, Inc; received funding for the conduct of clinical trials from Seattle Genetics; and served on an advisory board for Allakos, Inc. T.I.G. has served as a consultant for Novartis, Blueprint Medicines, Deciphera, Inc., and Allakos, Inc. A.R. has served on advisory boards for and received honoraria, travel reimbursement, and funding for the conduct of clinical trials from Novartis Pharma, Blueprint Medicines, and Deciphera, Inc.
Correspondence: Jason Gotlib, Stanford Cancer Institute, 875 Blake Wilbur Dr, Room 2324, Stanford, CA 94305-6555; e-mail: jason.gotlib@stanford.edu.
