

Introduction: Acetylated histone and non-histone proteins are pharmacologic targets for both solid and hematological cancers including myeloproliferative neoplasms (MPNs), a group of clonal hematological malignancies driven by aberrant JAK2/STAT signaling. MPNs are characterized by epigenetic alterations, including aberrant acetylation, which makes this disease particularly interesting for targeting with HDAC inhibitors. Four classes of histone deacetylases (Class I-IV HDACs) regulate gene transcription and modulate cellular processes that drive the initiation and progression of cancer. Pan-HDAC and class I-selective HDAC inhibitors have gained traction in clinical settings, yet we reasoned that specific targeting of the 18 distinct HDAC proteins may establish roles for select HDACs as therapeutic vulnerabilities in MPNs.
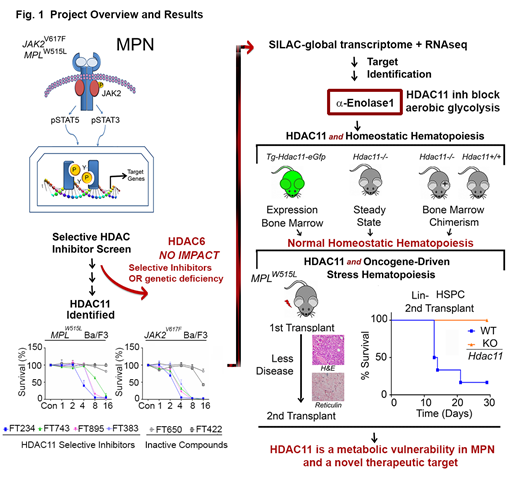
Methods: To explore the roles of individual HDACs in MPN, we first conducted an inhibitor screen of compounds having distinct HDAC selectivity based on electrophoretic mobility shift assays with full-length human HDAC proteins expressed in baculovirus and unique peptide substrates. Ultra-specific HDAC6 compounds were initially targeted for analysis based on its previously defined role in HSP90-mediated JAK2 stabilization and translation. Survival of MPN cell line models, MPN patient samples, leukemia cell lines, and MPN disease progression in mice transplanted with Hdac6-/-, and Hdac11-/- hematopoietic stem cells (HSCs) transduced with the MPLW515L oncogene, as well as Tg-Hdac11-eGfp mice were used to show the role of HDAC6 and HDAC11 in oncogene-driven and homeostatic hematopoiesis. As further proof of specificity, HDAC6 and HDAC11 were genetically ablated in MPN model cell lines using either RNA interference or inducible shRNA. For HDAC11 substrate identification, a combination of RNA-seq, acetylated proteome (SILAC), global metabolomics (LC-MS), Seahorse metabolic assays (Agilent Technologies), enzymatic assays, and acetylation-specific immunoblotting and mutation profiling were performed (Fig. 1).
Results: Despite the established interplay between HDAC6, HSP90 and JAK2, neither a highly selective HDAC6 inhibitor, HDAC6 silencing, nor the Hdac6 deficiency suppressed MPN pathogenesis, although there were clear effects on the acetylation of α-tubulin, a well characterized HDAC6-selective substrate. Intriguingly, both inhibition of HDAC11 activity with highly-specific HDAC11 inhibitors and silencing HDAC11 using an inducible validated shRNA, identified HDAC11 as a therapeutic vulnerability for multiple human MPN cell lines. The Tg-Hdac11-eGFP reporter mice showed that HDAC11 is expressed in several hematopoietic cell types, including myeloid cells, erythroblasts, and megakaryocytes. Thus, Hdac11-/- and Hdac11+/+MPLWT bone marrow were examined for steady-state hematopoiesis and transplantation chimerism. These studies demonstrated that HDAC11 does not contribute to homeostatic or transplantated bone marrow reconstitution. However, in the oncogenic MPL model, recipient mice transplanted withoncogenic MPLW515L-expressing Hdac11-deficient HSCs displayed markedly impaired cytokine-independent colony-formation, had less fibrosis, and displayed improved survival in primary and secondary MPN hematopoietic stem cell transplantation; thus HDAC11 contributes to MPN pathogenesis (Fig. 1). Studies in additional leukemia cell lines, including THP-1, HL-60, and mantle lymphoma cell lines, but not in Ramos or K562 cells, established that HDAC11 contributes to oncogene-driven events in other cell types. Mechanistically, RNA-seq, SILAC proteomics, and metabolic profiling revealed that HDAC11 controls aerobic glycolysis by deacetylating Lys343 of the glycolytic enzyme enolase-1 (ENO1), functionally inactivating ENO1. Finally, the effects of targeting HDAC11 on metabolism were augmented by blocking compensatory pathways of oxidative phosphorylation that are induced via JAK2V617Fand MPLW515L oncogenic signaling.
Conclusions: Our comprehensive screens of HDAC inhibitors, coupled with our biological, in vivo and molecular studies, indicate that HDAC11 is an attractive and potent target for disabling MPN metabolism and pathogenesis. These finding support the rationale for further development of clinical HDAC11 inhibitors for the treatment of metabolically-active cancers such as MPNs.
Pinilla Ibarz:Teva: Consultancy; TG Therapeutics: Consultancy; Sanofi: Speakers Bureau; Bayer: Speakers Bureau; Novartis: Consultancy; Bristol-Myers Squibb: Consultancy; Abbvie: Consultancy, Speakers Bureau; Takeda: Consultancy, Speakers Bureau; Janssen: Consultancy, Speakers Bureau. Reuther:Incyte Corporation: Research Funding. Levine:Loxo: Membership on an entity's Board of Directors or advisory committees; Roche: Consultancy, Research Funding; Lilly: Honoraria; C4 Therapeutics: Membership on an entity's Board of Directors or advisory committees; Isoplexis: Membership on an entity's Board of Directors or advisory committees; Imago Biosciences: Membership on an entity's Board of Directors or advisory committees; Novartis: Consultancy; Gilead: Consultancy; Celgene: Consultancy, Research Funding; Qiagen: Membership on an entity's Board of Directors or advisory committees; Prelude Therapeutics: Research Funding; Amgen: Honoraria. Verma:BMS: Research Funding; Janssen: Research Funding; Stelexis: Equity Ownership, Honoraria; Acceleron: Honoraria; Celgene: Honoraria. Epling-Burnette:Incyte Corporation: Research Funding; Celgene Corporation: Patents & Royalties, Research Funding; Forma Therapeutics: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal