

Key Points
UAE inhibition with TAK-243 induces an ER stress response and apoptosis in myeloma models.
TAK-243 overcomes drug resistance, and shows activity against primary and in vivo models, supporting its translation to the clinic.

Abstract
Three proteasome inhibitors have garnered regulatory approvals in various multiple myeloma settings; but drug resistance is an emerging challenge, prompting interest in blocking upstream components of the ubiquitin-proteasome pathway. One such attractive target is the E1 ubiquitin-activating enzyme (UAE); we therefore evaluated the activity of TAK-243, a novel and specific UAE inhibitor. TAK-243 potently suppressed myeloma cell line growth, induced apoptosis, and activated caspases while decreasing the abundance of ubiquitin-protein conjugates. This was accompanied by stabilization of many short-lived proteins, including p53, myeloid cell leukemia 1 (MCL-1), and c-MYC, and activation of the activating transcription factor 6 (ATF-6), inositol-requiring enzyme 1 (IRE-1), and protein kinase RNA-like endoplasmic reticulum (ER) kinase (PERK) arms of the ER stress response pathway, as well as oxidative stress. UAE inhibition showed comparable activity against otherwise isogenic cell lines with wild-type (WT) or deleted p53 despite induction of TP53 signaling in WT cells. Notably, TAK-243 overcame resistance to conventional drugs and novel agents in cell-line models, including bortezomib and carfilzomib resistance, and showed activity against primary cells from relapsed/refractory myeloma patients. In addition, TAK-243 showed strong synergy with a number of antimyeloma agents, including doxorubicin, melphalan, and panobinostat as measured by low combination indices. Finally, TAK-243 was active against a number of in vivo myeloma models in association with activation of ER stress. Taken together, the data support the conclusion that UAE inhibition could be an attractive strategy to move forward to the clinic for patients with relapsed and/or refractory multiple myeloma.
Introduction
Outcomes for multiple myeloma patients have improved significantly with the introduction of novel agents,1 but the majority suffer multiple relapses characterized by shorter durations of clinical benefit with each line of therapy.2 This was recently underscored by a study evaluating outcomes in patients with immunomodulatory agent (immunomodulatory drug [IMiD])–refractory and proteasome inhibitor (PI)–refractory disease, whose median survival was only 13 months.3 These outcomes will be improved further by deacetylase inhibitors such as panobinostat,4 and monoclonal antibodies such as daratumumab5-7 and elotuzumab.8 Integration into our treatment approaches of the latter class especially will hopefully provide dramatic benefits.9 Unfortunately, even these agents have decreased efficacy in patients with quadruple-refractory disease,10 defined as myeloma that has progressed despite 2 PIs and 2 IMiDs. Thus, a group of patients with relapsed/refractory disease who could benefit from agents with new mechanisms of action can still be identified, especially if they overcome novel and conventional drug resistance.
One of the most successful approaches to myeloma therapy has been through ubiquitin-proteasome pathway (UPP) inhibition.11 Three PIs have received regulatory approval, including the reversible inhibitors bortezomib and ixazomib, and the irreversible carfilzomib. These function in part by disturbing the balance between proteasome load and capacity12 and the unfolded protein response (UPR).13-15 Early UPR events reduce endoplasmic reticulum (ER) stress by increasing protein-folding capacity and reducing ER protein load.16 This occurs through 3 signaling arms that involve protein kinase RNA-like ER kinase (PERK), inositol-requiring enzyme 1α (IRE-1α), and activating transcription factor 6 (ATF-6).16 Indeed, the UPR’s importance is underscored by findings suggesting that PI-resistance mechanisms enhance proteasome capacity or reduce proteasome load.17-19 Such adaptations restore the balance between capacity and load, thereby reducing ER stress and reliance on the UPR.11 Prolonged UPR induction, however, results in activation of a proapoptotic, terminal UPR phase.16
The proteasome represents the UPP’s final common effector that digests proteins intended for turnover, but this pathway has other targets. E3 ubiquitin ligases such as cereblon20 and murine double minute 2 (MDM-2),21-24 which ubiquitinate a small subset of client proteins and could provide greater target specificity, represent one example. Deubiquitinases, which remove ubiquitin chains from proteins, are also promising targets.25-29 Even further upstream is the E1 ubiquitin-activating enzyme (UAE), which activates ubiquitin in an adenosine triphosphate (ATP)-dependent fashion to allow its later transfer to target proteins.30 Knockdown of E1 using short hairpin RNAs (shRNAs) produced cytotoxic effects in leukemia and myeloma cell line models, and an E1 inhibitor tool compound reduced viability as well,31 providing impetus to study this target. In the current report, we evaluated the efficacy and mechanisms of action of TAK-243, a physiologically relevant E1 inhibitor32,33 undergoing clinical testing. In addition to single-agent activity against drug-naive myeloma cell lines in vitro and in vivo, and against primary patient-derived cells, TAK-243 overcame resistance to conventional and novel drugs. Mechanistically, TAK-243 induced apoptosis and UPR dysregulation, with greater potency in some cases than bortezomib. Finally, it showed synergistic interactions with some drugs that are part of our therapeutic armamentarium, suggesting that it may hold promise to provide another option for relapsed/refractory myeloma patients.
Methods
Reagents
TAK-243 was obtained from Millennium Pharmaceuticals, Inc, a subsidiary of Takeda Pharmaceutical Company Limited (Cambridge, MA) (see supplemental Methods [available on the Blood Web site] for additional details about the sources of the reagents used).
Myeloma cell lines and primary samples
Drug-resistant myeloma cells and isogenic cell lines with wild-type (WT) or deleted p53 were developed and maintained as described previously.34 The pZsProSensor-1 Vector (Clontech Laboratories, Inc, Mountain View, CA) expressing the ZsGreen-MODC-d410 fusion protein, which is degraded by the proteasome in a ubiquitin-independent manner,35 was stably transfected into myeloma cells (see supplemental Methods for additional details).
Cell-viability assays
Cell lines were treated with the indicated agents for 24 hours, and for 72 hours in synergy experiments and with primary samples. This was followed by addition of the WST-1 tetrazolium reagent (Roche Applied Science, Indianapolis, IN); colorimetric detection of metabolic activity was performed on a Victor3V plate reader (PerkinElmer Life Sciences, Boston, MA). Viability data were normalized to vehicle controls set at 100%, and data points were represented as the mean with the standard deviation (SD).
Flow cytometry
Cell apoptosis was measured after Annexin V Pacific Blue and TO-PRO-3 (Invitrogen) staining using a BD FACSCANTO II flow cytometer and FlowJo Version 7.6.1.36 Caspases 3, 8, and 9 were detected by flow cytometry after staining with sulforhodamine-labeled inhibitors from the CaspGLOW Red Active Staining kit (BioVision, Inc, Milpitas, CA). Expression of the pZsProSensor-1 vector was detected by flow as a green fluorescence.
Real-time reverse transcription–PCR
Real-time quantitative polymerase chain reaction (PCR; qPCR) was carried out as described previously37 (see supplemental Methods for additional details).
Western blotting
Harvested cells were lysed using 1× lysis buffer (Cell Signaling Technology, Danvers, MA) as described previously19 (see supplemental Methods for additional details).
Gene expression profiling
Total RNA was extracted from cells, and 300 ng was amplified and biotin-labeled using an Eberwine procedure in an Illumina TotalPrep RNA amplification kit (Thermo Fisher Scientific). The RNA was hybridized to Illumina HT12 version 4 human whole-genome microarrays, and processing of bead-level data were as previously described.38 Significance testing for differentially expressed probes was by the Wilcoxon rank-sum test applied to individual processed bead values, with false-discovery rate significance values (q) determined by the method of Hochberg and Benjamini.39
Reverse-phase protein array
Two million MM1.S or U266 myeloma cells were treated with TAK-243 at the indicated concentrations for 24 hours. Drug-naive and -treated cells were harvested and submitted for reverse-phase protein array (RPPA) to measure protein changes across a set of antibodies through our RPPA Core Facility, and data were analyzed as described previously.40,41
Drug-synergy calculations
Synergy experiments were carried out as previously described.42 In brief, TAK-243, lenalidomide, panobinostat, melphalan, or doxorubicin was added to U266 cells, and the median inhibitor concentration (IC50) of each drug individually was determined. A range of serial dilutions was made across the IC50 dose range, with the IC50 set as 1×, and dilutions were made relative to this value. The agents were then added simultaneously for 72 hours, and WST-1 assays were performed. Data were analyzed using CalcuSyn software (Biosoft, Cambridge, United Kingdom). Combination indices (CIs) were calculated, and values <1.0 were considered to indicate synergy.
Tumor xenografts
CB-17 severe combined immunodeficient (SCID) mice (Charles River Laboratories, Wilmington, MA) were inoculated subcutaneously in the flanks with MM1.S or MOLP-8 cells in RPMI 1640 media with Matrigel. Tumor growth was monitored with Vernier calipers, and mean tumor volume was calculated as described in supplemental Methods, where details are also provided about the pharmacodynamics studies performed.
Results
UAE inhibition and myeloma cell viability
To determine the potential of UAE inhibition in myeloma, we exposed a panel of cell lines to TAK-243, a physiologically relevant E1 inhibitor32,33 (supplemental Figure 1), for 24 hours. Most were quite sensitive, with an IC50 of 25 to 100 nM (Figure 1A), such as MM1.S cells, where this value was 25 nM. Comparable sensitivity to TAK-243 was seen in B-cell lymphoma cell lines, whereas epithelial cancer cells and nontransformed cells were generally less sensitive (supplemental Figure 2A). Two myeloma lines that were less sensitive were U266 and RPMI 8226 cells, which had an IC50 of 250 and >1000 nM, respectively. Based on staining with Annexin V and TO-PRO-3, MM1.S cells exposed to TAK-243 revealed an increase in early (Annexin V+/TO-PRO-3−) and late (Annexin V+/TO-PRO-3+) apoptosis (supplemental Figure 2B) in a dose-dependent manner (Figure 1B). This was associated with activation of initiator caspases, including caspases-8 and -9, and the executioner caspase-3 (Figure 1C), as was the case for bortezomib, and a pan-caspase inhibitor reduced apoptosis (Figure 1B). Caspase-7 was another executioner caspase activated in MM1.S cells (Figure 1D), and this could also be detected with higher drug concentrations in U266 cells (supplemental Figure 2C). Notably, UAE knockdown with shRNAs (supplemental Figure 3A) reduced cell sensitivity to TAK-243 (supplemental Figure 3B) in MM1.S and U266 cells.
![Figure 1. TAK-243 reduces cell viability and induces apoptosis. (A) A panel of myeloma cell lines was exposed to either vehicle (dimethyl sulfoxide [DMSO]) or TAK-243 at the indicated nanomolar concentrations, and viability was then determined and plotted. (B) Apoptosis was evaluated after exposure to TAK-243 in MM1.S cells by staining with Annexin V and TO-PRO-3, with bortezomib (BTZ) at 10 nM as a positive control, and in the presence of TAK-243 with the pan-caspase inhibitor Z-VAD-FMK. (C) Activation of caspases-8, -9, and -3 was determined after TAK-243 exposure using fluorogenic substrates, and normalized to the vehicle control, which was arbitrarily set at 1.0. Data represent 3 independent experiments, each performed in triplicate, and are presented as the mean plus or minus SD; *P < .05 compared with the respective controls in this and panels of later figures. (D) Cleavage of caspase-7 was verified by western blotting of MM1.S cell extracts after exposure to the indicated agents. Cl. Caspase, cleaved caspase.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/14/10.1182_blood-2018-06-859686/4/m_blood859686f1.png?Expires=1769090239&Signature=CG34G0DDhXFGQrPJulUNogUBofYUGakMl5TiydEnx6K7Xz0xxvfaktmLlMwuCR1tCSmcTe~m6YmHOdvkKx0QLU70zOjNecNoTNdspoyEXY5jzZvPv-FQ97QqmHtdac0kkCq4pBKSyhlTHiUT8rkL1cVyeMGl8QysIRWfhNNHjoC7LGSxRIx4Y~ChNonL7B3n-Cnk9MKEMPHGJGsVRPKYyOLZgafNkmGti-75AeHOe1YvdWYGD5~buBhmo5HAG~Fga811DwBYTXYFPKglSeqWpxTKYqgy7Gntz1QzRi4OAiIGeyVTXeKtp0uESogICVY75Us4yl5lqmv9TUi0BtzfTQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
TAK-243 reduces cell viability and induces apoptosis. (A) A panel of myeloma cell lines was exposed to either vehicle (dimethyl sulfoxide [DMSO]) or TAK-243 at the indicated nanomolar concentrations, and viability was then determined and plotted. (B) Apoptosis was evaluated after exposure to TAK-243 in MM1.S cells by staining with Annexin V and TO-PRO-3, with bortezomib (BTZ) at 10 nM as a positive control, and in the presence of TAK-243 with the pan-caspase inhibitor Z-VAD-FMK. (C) Activation of caspases-8, -9, and -3 was determined after TAK-243 exposure using fluorogenic substrates, and normalized to the vehicle control, which was arbitrarily set at 1.0. Data represent 3 independent experiments, each performed in triplicate, and are presented as the mean plus or minus SD; *P < .05 compared with the respective controls in this and panels of later figures. (D) Cleavage of caspase-7 was verified by western blotting of MM1.S cell extracts after exposure to the indicated agents. Cl. Caspase, cleaved caspase.
TAK-243 reduces cell viability and induces apoptosis. (A) A panel of myeloma cell lines was exposed to either vehicle (dimethyl sulfoxide [DMSO]) or TAK-243 at the indicated nanomolar concentrations, and viability was then determined and plotted. (B) Apoptosis was evaluated after exposure to TAK-243 in MM1.S cells by staining with Annexin V and TO-PRO-3, with bortezomib (BTZ) at 10 nM as a positive control, and in the presence of TAK-243 with the pan-caspase inhibitor Z-VAD-FMK. (C) Activation of caspases-8, -9, and -3 was determined after TAK-243 exposure using fluorogenic substrates, and normalized to the vehicle control, which was arbitrarily set at 1.0. Data represent 3 independent experiments, each performed in triplicate, and are presented as the mean plus or minus SD; *P < .05 compared with the respective controls in this and panels of later figures. (D) Cleavage of caspase-7 was verified by western blotting of MM1.S cell extracts after exposure to the indicated agents. Cl. Caspase, cleaved caspase.
TAK-243 does not block ubiquitin-independent proteolysis
Agents like bortezomib suppress turnover of proteins degraded through the proteasome in a ubiquitin-independent or ubiquitin-dependent manner. The latter was illustrated by an accumulation of ubiquitin-protein conjugates in bortezomib-exposed MM1.S (Figure 2A left panel) or U266 cells (Figure 2A right). In contrast, TAK-243 did not increase ubiquitin-protein conjugates and, if anything, reduced them (Figure 2A). Similarly, the combination of bortezomib and TAK-243 led to reduced conjugate levels, as would be expected because ubiquitin activation is necessary for its transfer to target protein lysine residues.11 To further probe TAK-243’s impact on proteolysis, we overexpressed the Zoanthus sp. green fluorescent protein (ZsGreen) fused to the mouse ornithine decarboxylase degradation (MODC) domain, which is degraded in a proteasome-dependent but ubiquitin-independent manner.43 Although bortezomib enhanced ZsGreen-MODS abundance in ANBL-6 and RPMI 8226 cells (Figure 2B), this was not the case for TAK-243, consistent with a mechanism that did not interfere with proteasome activity. In contrast, myeloid cell leukemia 1 (MCL-1) and c-MYC, which are degraded in a ubiquitin- and proteasome-dependent manner, did accumulate in the presence of TAK-243 or bortezomib (Figure 2C).
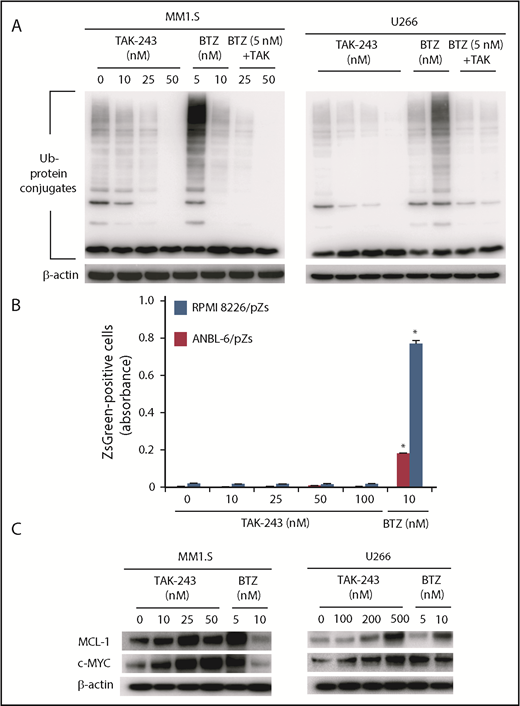
UAE inhibition does not impact upon proteasome function. (A) Accumulation of ubiquitin (Ub)-protein conjugates was evaluated in the indicated cell lines after exposure to TAK-243, bortezomib, or to the combination of the 2 agents at the indicated concentrations for 24 hours. (B) RPMI 8226 and ANBL-6 cells transfected with a vector expressing the ZsGreen-MODC-d410 fusion protein were exposed to TAK-243 or bortezomib at the indicated concentrations. Expression levels of this protein, which is degraded in a proteasome-dependent but ubiquitin-independent manner, were then evaluated by fluorescence monitoring, and the panel shows fluorescence (λex 488 nM/λem 510 nM). (C) MM1.S and U266 cells were then exposed to TAK-243 or bortezomib under the conditions described in panel A, and western blotting was used to determine the abundance of MCL1 and cMYC.
UAE inhibition does not impact upon proteasome function. (A) Accumulation of ubiquitin (Ub)-protein conjugates was evaluated in the indicated cell lines after exposure to TAK-243, bortezomib, or to the combination of the 2 agents at the indicated concentrations for 24 hours. (B) RPMI 8226 and ANBL-6 cells transfected with a vector expressing the ZsGreen-MODC-d410 fusion protein were exposed to TAK-243 or bortezomib at the indicated concentrations. Expression levels of this protein, which is degraded in a proteasome-dependent but ubiquitin-independent manner, were then evaluated by fluorescence monitoring, and the panel shows fluorescence (λex 488 nM/λem 510 nM). (C) MM1.S and U266 cells were then exposed to TAK-243 or bortezomib under the conditions described in panel A, and western blotting was used to determine the abundance of MCL1 and cMYC.
Impact of TAK-243 on the proteome
To gain a broader understanding of the effect of TAK-243 on myeloma cells, we performed RPPA analysis on vehicle- and TAK-243–treated MM1.S (Figure 3A; supplemental Figure 4) and U266 (supplemental Figures 5 and 6) cells. Consistent with the induction of caspase-mediated cell death, cleaved caspase-3 and -7 increased in MM1.S (Figure 3A) and U266 (supplemental Figure 6) cells, as did phospho–c-Jun–N-terminal kinase and BCL-2–interacting mediator of cell death (BIM). TAK-243 enhanced the abundance of a number of short-lived proteins whose turnover occurs through ubiquitin- and proteasome-dependent proteolysis, including MCL1 and hypoxia-inducible factor 1α (HIF-1α) in both cell lines, and c-MYC in MM1.S cells (Figure 3B; supplemental Table 1). Caspase inhibition increased the abundance of both MCL1 and c-MYC (Figure 3B), supporting the likelihood that activation of apoptosis may impact upon the abundance of some cellular proteins under these conditions. As MM1.S harbor a WT p53, increased levels of p53 and some of its downstream targets were seen, including MDM-2 and p21. Finally, evidence was seen that TAK-243 activated a stress response due to an increase in heat shock protein 70 (HSP-70), the oxidative stress gene superoxide dismutase 2 (SOD-2), and Tu translation elongation factor, mitochondrial (TUFM) expression. Gene expression profiling was performed on treated MM1.S cells as well (Figure 3C; Gene Expression Omnibus [GEO] accession number GSE126254). Significantly enriched gene sets with a false discovery rate of <5% are presented in Table 1 as hallmark gene sets.44
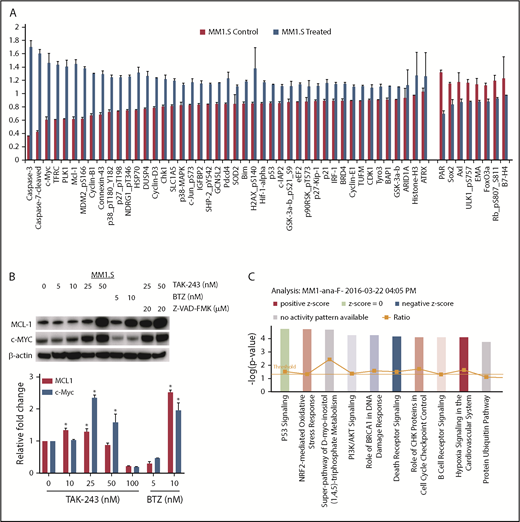
TAK-243 impacts on protein and gene expression profiles. (A) RPPA analysis was performed on MM1.S cells exposed to vehicle or TAK-243. The 45 proteins whose abundance was most enhanced are indicated at the left, whereas the right side shows the 8 whose abundance was most decreased. (B) Western blotting (top panel) was performed to confirm the changes in MCL-1 and c-MYC, with β-actin serving as a loading control. Bortezomib serves as a positive control, and studies were also performed in the presence of Z-VAD-FMK to determine a possible impact of activation of apoptosis on the abundance of these proteins. Changes in protein-expression levels were largely independent of any alterations in the abundance of the respective messenger RNAs (bottom panel). (C) Gene expression profiling was performed on TAK-243–treated cells, and the top 10 dysregulated pathways identified by Ingenuity pathway analysis are indicated.
TAK-243 impacts on protein and gene expression profiles. (A) RPPA analysis was performed on MM1.S cells exposed to vehicle or TAK-243. The 45 proteins whose abundance was most enhanced are indicated at the left, whereas the right side shows the 8 whose abundance was most decreased. (B) Western blotting (top panel) was performed to confirm the changes in MCL-1 and c-MYC, with β-actin serving as a loading control. Bortezomib serves as a positive control, and studies were also performed in the presence of Z-VAD-FMK to determine a possible impact of activation of apoptosis on the abundance of these proteins. Changes in protein-expression levels were largely independent of any alterations in the abundance of the respective messenger RNAs (bottom panel). (C) Gene expression profiling was performed on TAK-243–treated cells, and the top 10 dysregulated pathways identified by Ingenuity pathway analysis are indicated.
Hallmark gene sets identified from the Molecular Signatures Database using gene expression data from MM1.S cells exposed to TAK-243
| Gene sets |
|---|
| Upregulated |
| HALLMARK_TNFA_SIGNALING_VIA_NFκB |
| HALLMARK_UNFOLDED_PROTEIN_RESPONSE |
| HALLMARK_P53_PATHWAY |
| HALLMARK_APOPTOSIS |
| HALLMARK_INFLAMMATORY_RESPONSE |
| HALLMARK_HYPOXIA |
| HALLMARK_MYC_TARGETS_V2 |
| Downregulated |
| HALLMARK_PEROXISOME |
| HALLMARK_BILE_ACID_METABOLISM |
| HALLMARK_E2F_TARGETS |
| HALLMARK_FATTY_ACID_METABOLISM |
| HALLMARK_OXIDATIVE_PHOSPHORYLATION |
| HALLMARK_HEME_METABOLISM |
| Gene sets |
|---|
| Upregulated |
| HALLMARK_TNFA_SIGNALING_VIA_NFκB |
| HALLMARK_UNFOLDED_PROTEIN_RESPONSE |
| HALLMARK_P53_PATHWAY |
| HALLMARK_APOPTOSIS |
| HALLMARK_INFLAMMATORY_RESPONSE |
| HALLMARK_HYPOXIA |
| HALLMARK_MYC_TARGETS_V2 |
| Downregulated |
| HALLMARK_PEROXISOME |
| HALLMARK_BILE_ACID_METABOLISM |
| HALLMARK_E2F_TARGETS |
| HALLMARK_FATTY_ACID_METABOLISM |
| HALLMARK_OXIDATIVE_PHOSPHORYLATION |
| HALLMARK_HEME_METABOLISM |
TAK-243 enhanced the expression of UPR-relevant proteins and genes
As PIs induce cell death in part by activating a terminal UPR,14,15 and both gene expression profiling and RPPA data supported that TAK-243 also induced stress pathways, we looked in more detail at the UPR components. Notably, TAK-243 activated 3 UPR16 arms in MM1.S (Figure 4A left panel) and U266 (Figure 4A right) cells. This was evidenced by increased PERK phosphorylation, and increased ATF-6 and XBP1s expression, the latter of which is downstream of IRE-1α. Interestingly, in some cases, this activation was equal to or stronger than that with bortezomib at comparable drug concentrations, such as of ATF6. Additional notable changes included increased ATF4, Binding immunoglobulin protein (BiP), C/EBP homologous protein (CHOP), HSP70, and NRF2, consistent with ER stress induction. These same proteins were modulated by shRNA-mediated knockdown of UAE (supplemental Figure 3C). qPCR studies confirmed induction of gene expression for many of these UPR components by TAK-243, especially in MM1.S (Figure 4B top panel), and also to some extent in U266 cells (Figure 4B bottom). The maximal changes of messenger RNA expression were at 50 and 200 nM in MM1.S and U266 cells, respectively, consistent with their IC50 values.
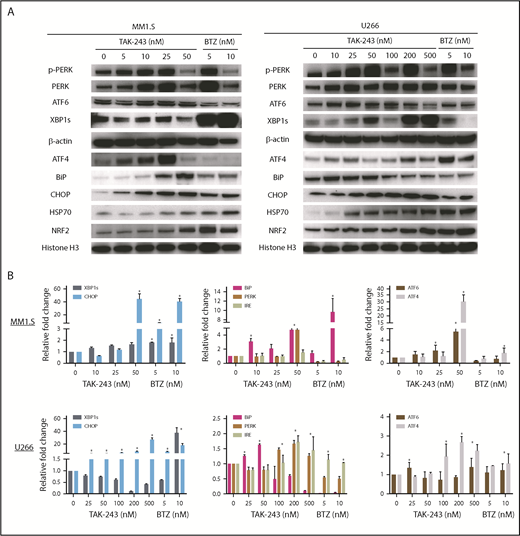
TAK-243 activates stress-response pathways. (A) The impact of UAE inhibition on selected components of the stress-response pathway is shown by western blotting of extracts from MM1.S and U266 myeloma cells, with bortezomib serving as a positive control. Loading was confirmed using either β-actin or histone H3 as an internal control. (B) qPCR was also performed to evaluate any changes in expression of stress-response pathway genes under the same conditions.
TAK-243 activates stress-response pathways. (A) The impact of UAE inhibition on selected components of the stress-response pathway is shown by western blotting of extracts from MM1.S and U266 myeloma cells, with bortezomib serving as a positive control. Loading was confirmed using either β-actin or histone H3 as an internal control. (B) qPCR was also performed to evaluate any changes in expression of stress-response pathway genes under the same conditions.
UAE inhibition and p53 signaling
Activation of p53 signaling seen by RPPA was next evaluated in MM1.S and U266 cells by western blotting. TAK-243 induced accumulation of WT p53 and its downstream targets MDM2 and p21 in MM1.S cells (Figure 5A left panel), but had no discernible effect on p53 and MDM2 in p53-mutant U266 cells (Figure 5A right). p53 loss represents a high-risk feature in myeloma, and identifies a population for whom novel therapies are needed.45 Our earlier data showed that the IC50 was higher for TAK-243 in U266 cells, suggesting that this agent worked in a p53-dependent fashion. However, to test this more thoroughly, we compared otherwise isogenic cells that, through genome editing, harbored a WT or deleted p53.34 Interestingly, a much more modest IC50 difference was seen comparing MM1.S WT and knockout cells (Figure 5B left panel), at 16.8 and 27.1 nM, respectively, whereas no difference was seen comparing MOLP-8 WT and knockout (KO) cells (27.1 and 21.6 nM, respectively) (Figure 5B right). Western blotting confirmed the induction of a p53-dependent cascade in the WT cells (Figure 5C), including p53 itself, MDM2, and p21, whereas p21 induction was also seen, albeit to a lesser extent, in the KOs. Also of note, BIM levels were modestly increased in TP53 WT and KO models consistent with the RPPA data in Figure 3A, and could provide a further proapoptotic stimulus.

Impact of TAK-243 on the p53 pathway. (A) Accumulation of p53 pathway–related proteins is demonstrated in p53 WT MM1.S cells (left panel) by western blotting, but not in TP53 mutant U266 cells (right). (B) Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas-9) methodology was used to generate MM1.S cells in which p53 expression was knocked out, or cells in which a nontargeting (NT) construct was added. These were then compared for their sensitivity to TAK-243 by measuring viability with a tetrazolium-based assay (left panel). MOLP-8 cells in which p53 had been knocked out using sequence-specific zinc finger nucleases (ZFNs) were similarly compared with MOLP-8 cells with a nontargeting ZFN (right panel). Data are presented as the mean plus or minus SD of triplicate experiments; *P < .05 when compared with controls. (C) The impact of TAK-243 on selected p53 pathway and stress-response proteins was evaluated in these p53 WT and KO cells by western blotting.
Impact of TAK-243 on the p53 pathway. (A) Accumulation of p53 pathway–related proteins is demonstrated in p53 WT MM1.S cells (left panel) by western blotting, but not in TP53 mutant U266 cells (right). (B) Clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas-9) methodology was used to generate MM1.S cells in which p53 expression was knocked out, or cells in which a nontargeting (NT) construct was added. These were then compared for their sensitivity to TAK-243 by measuring viability with a tetrazolium-based assay (left panel). MOLP-8 cells in which p53 had been knocked out using sequence-specific zinc finger nucleases (ZFNs) were similarly compared with MOLP-8 cells with a nontargeting ZFN (right panel). Data are presented as the mean plus or minus SD of triplicate experiments; *P < .05 when compared with controls. (C) The impact of TAK-243 on selected p53 pathway and stress-response proteins was evaluated in these p53 WT and KO cells by western blotting.
Activity of TAK-243 on primary and drug-resistant myeloma cells
To evaluate whether TAK-243 was active against primary cells, we examined its efficacy against freshly isolated CD138+ plasma cells from myeloma patients. After a 72-hour exposure to TAK-243 in 8 unique primary samples, all showed a substantial reduction in viability (Figure 6A left panel), with an IC50 of 50 to 200 nM. Studies of the induction of apoptosis were performed by flow cytometry in 3 samples for which we had sufficient cells after Annexin V and TO-PRO-3 staining. The majority were Annexin V+/TO-PRO-3− (Figure 6A right panel), consistent with their entry into early apoptosis, though an increase was also seen in late (Annexin V+/TO-PRO-3+) apoptotic cells. As patients in the refractory setting often have drug-resistant disease, we did look at a number of such models, including resistance to conventional drugs like dexamethasone, doxorubicin, and melphalan. RPMI 8226 WT cells were not very sensitive to TAK-243 (IC50 1.75 μM), as indicated earlier, but doxorubicin-resistant RPMI 8226/DOX40 and melphalan-resistant RPMI 8226/LR5 cells were more sensitive (IC50 of 1.50 and 1.05 μM, respectively) to UAE inhibition (Figure 6B; supplemental Table 2). Similarly, MM1.R dexamethasone-resistant cells showed a comparable sensitivity to MM1.S corticosteroid-sensitive cells. With regard to novel agents, we also evaluated TAK-243 in MM1.S lenalidomide-resistant (R10R) cells, and in ANBL-6 bortezomib-resistant (V10R) cells. Compared with their WT counterparts, only small differences were seen in the IC50 values (24 vs 20 nM for the lenalidomide-sensitive and -resistant cells, and 10.9 vs 6.4 nM for the bortezomib-sensitive and -resistant cells, respectively) (Figure 6B). Similar findings were noted in carfilzomib-resistant KAS-6/1 cells, which had an IC50 of 4.9 nM compared with 7.4 nM for their WT counterparts (supplemental Table 2). Furthermore, studies in ANBL-6 drug-naive and V10R and C10R cells also showed, if anything, a trend toward greater sensitivity for the drug-resistant cells to TAK-243 (supplemental Table 2). Interestingly, when TAK-243 was added to bortezomib or carfilzomib in either WT or PI-resistant cells, enhanced activity of the combinations was not seen (supplemental Figure 7). Indeed, CI analysis revealed findings consistent with antagonism between the 2 sets of agents targeting the UPP (supplemental Table 3).

UAE inhibition overcomes drug resistance and enhances chemosensitivity. (A) Bone marrow plasma cells were purified from 8 previously treated myeloma patients, exposed to different doses of TAK-243 for 72 hours, and their viability was evaluated using a tetrazolium-based assay. (B) A panel of drug-resistant myeloma cell lines was compared with their drug-sensitive controls for their susceptibility to TAK-243. The panel included doxorubicin-resistant RPMI 8226/DOX 40 cells, melphalan-resistant RPMI 8226/LR-5 cells, lenalidomide-resistant MM1.S/R10R cells, dexamethasone-resistant MM1.R cells, and bortezomib-resistant KAS-6/1 V10R cells. (C) TAK-243 alone (at the indicated concentrations on the top axes), and either panobinostat (left panel) alone (PAN; at the indicated concentrations on the bottom axis) or lenalidomide (right panel) alone (LEN; at the indicated concentrations on the bottom axis) were added to U266 cells. Also, TAK-243 at the concentrations indicated in the legend was combined with either panobinostat or lenalidomide at the drug concentrations indicated in the lower abscissas to determine whether additive or synergistic interactions were seen during 72-hour drug exposures.
UAE inhibition overcomes drug resistance and enhances chemosensitivity. (A) Bone marrow plasma cells were purified from 8 previously treated myeloma patients, exposed to different doses of TAK-243 for 72 hours, and their viability was evaluated using a tetrazolium-based assay. (B) A panel of drug-resistant myeloma cell lines was compared with their drug-sensitive controls for their susceptibility to TAK-243. The panel included doxorubicin-resistant RPMI 8226/DOX 40 cells, melphalan-resistant RPMI 8226/LR-5 cells, lenalidomide-resistant MM1.S/R10R cells, dexamethasone-resistant MM1.R cells, and bortezomib-resistant KAS-6/1 V10R cells. (C) TAK-243 alone (at the indicated concentrations on the top axes), and either panobinostat (left panel) alone (PAN; at the indicated concentrations on the bottom axis) or lenalidomide (right panel) alone (LEN; at the indicated concentrations on the bottom axis) were added to U266 cells. Also, TAK-243 at the concentrations indicated in the legend was combined with either panobinostat or lenalidomide at the drug concentrations indicated in the lower abscissas to determine whether additive or synergistic interactions were seen during 72-hour drug exposures.
The majority of novel agents active against myeloma are used with other drugs to maximize their efficacy, and we evaluated a number of combinations. Because there is synergy between PIs and deacetylase inhibitors and IMiDs, we evaluated TAK-243 with panobinostat and lenalidomide. Addition of panobinostat at physiologically relevant concentrations to TAK-243 enhanced the antiproliferative effects of either agent alone in U266 cells (Figure 6C left panel), and CI analysis showed strong synergy (Table 2). In contrast, lenalidomide with TAK-243 produced antagonistic effects in U266 cells (Figure 6C right panel), though synergy was seen in KAS-6/1 cells (supplemental Table 4). Finally, because DNA damage repair pathways were impacted by TAK-243, we evaluated TAK-243 with melphalan or doxorubicin, and found the combinations reduced viability to a greater extent (supplemental Figure 8) and were synergistic (Table 2).
CI values of synergy experiments in U266 cells
| TAK-243 | ||||
|---|---|---|---|---|
| 25 nM | 50 nM | 100 nM | 200 nM | |
| Panobinostat, nM | ||||
| 3.125 | 0.26 | 0.256 | 0.297 | |
| 6.25 | 0.447 | 0.37 | 0.323 | |
| 12.5 | 0.513 | 0.521 | 0.465 | |
| Melphalan, nM | ||||
| 250 | 0.3127 | 0.2568 | 0.4683 | |
| 500 | 0.3496 | 0.3852 | 0.4748 | |
| 1000 | 0.3770 | 0.4826 | 0.5864 | |
| Doxorubicin, nM | ||||
| 62.5 | 0.3762 | 0.3582 | 0.4605 | |
| 125 | 0.5397 | 0.4085 | 0.613 | |
| 250 | 0.7009 | 0.6571 | 0.7471 | |
| TAK-243 | ||||
|---|---|---|---|---|
| 25 nM | 50 nM | 100 nM | 200 nM | |
| Panobinostat, nM | ||||
| 3.125 | 0.26 | 0.256 | 0.297 | |
| 6.25 | 0.447 | 0.37 | 0.323 | |
| 12.5 | 0.513 | 0.521 | 0.465 | |
| Melphalan, nM | ||||
| 250 | 0.3127 | 0.2568 | 0.4683 | |
| 500 | 0.3496 | 0.3852 | 0.4748 | |
| 1000 | 0.3770 | 0.4826 | 0.5864 | |
| Doxorubicin, nM | ||||
| 62.5 | 0.3762 | 0.3582 | 0.4605 | |
| 125 | 0.5397 | 0.4085 | 0.613 | |
| 250 | 0.7009 | 0.6571 | 0.7471 | |
UAE inhibition and in vivo antimyeloma activity
As a last evaluation of the potential of TAK-243, we studied its activity in xenograft models prepared using MM1.S or MOLP-8 cells. These were treated with vehicle, or TAK-243 at 12.5 mg/kg IV, or at 25 mg/kg IV, twice-weekly for 2 weeks. Twice-weekly dosing at 12.5 mg/kg produced tumor growth inhibition of 60% and 73% in the MM1.S and MOLP-8 models at 14 days (Figure 7A left and right panels, respectively). Dosing at 25 mg/kg gave an even greater impact, with an initial decline in tumor size in both models, followed by slowing of tumor progression. Western blotting of tumor tissue from the MM1.S xenografts treated with a single TAK-243 dose at 25 mg/kg showed a time-dependent increase in Noxa (Figure 7B), a proapoptotic, BH3-only protein that is degraded in a ubiquitin- and proteasome-dependent manner. Moreover, TAK-243 induced apoptosis in the MM1.S xenografts, as indicated by an increase in tumor cell–cleaved caspase-3 and cleaved poly-(ADP-ribose) polymerase. Finally, UAE inhibition in vivo activated similar mechanisms as had been shown in vitro (Figure 7C) because immunohistochemistry of MM1.S xenograft tissues showed increased XBP1s (supplemental Figure 9), BiP, and ATF4 (supplemental Figure 10) staining, confirming UPR activation.

Antitumor activity of TAK-243. (A) Xenografts based on either MM1.S (left panel) or MOLP-8 cells (right) were treated with either vehicle, or TAK-243 at a dose of either 12.5 mg/kg given twice weekly (BIW), or 25 mg/kg given once weekly (QW), and growth of subcutaneous tumors was monitored. (B) Expression levels of selected proteins of interest were determined by western blotting of dissected tumor tissues at the indicated time points after TAK-243 treatment. (C) Immunohistochemistry was also performed for selected proteins in tumor tissue in a separate cohort of mice from 1 to 24 hours after 1 dose treatment of TAK-243, and compared to vehicle-treated controls at 24 hours (hatched bars at left). PARP, poly-(ADP-ribose) polymerase.
Antitumor activity of TAK-243. (A) Xenografts based on either MM1.S (left panel) or MOLP-8 cells (right) were treated with either vehicle, or TAK-243 at a dose of either 12.5 mg/kg given twice weekly (BIW), or 25 mg/kg given once weekly (QW), and growth of subcutaneous tumors was monitored. (B) Expression levels of selected proteins of interest were determined by western blotting of dissected tumor tissues at the indicated time points after TAK-243 treatment. (C) Immunohistochemistry was also performed for selected proteins in tumor tissue in a separate cohort of mice from 1 to 24 hours after 1 dose treatment of TAK-243, and compared to vehicle-treated controls at 24 hours (hatched bars at left). PARP, poly-(ADP-ribose) polymerase.
Discussion
PIs suppress UPP function by binding to threonine residues of the catalytically active constitutive and immunoproteasome subunits, which are responsible for the nucleophilic attacks that break peptide bonds.11 In that 3 such drugs have received regulatory approval for myeloma patients and, in the case of bortezomib, for mantle cell lymphoma, there has been interest in studying upstream UPP targets. Although the proteasome is the final common effector for proteolysis through this pathway, our current study supports the possibility that inhibiting the very first step in this pathway, that of ubiquitin activation, could be a rational approach as well. Indeed, TAK-243 reduced myeloma cell viability and induced apoptosis (Figure 1), impacted ubiquitin-dependent but not ubiquitin-independent proteolysis (Figure 2), and induced the UPR in vitro and in vivo (Figures 4 and 7). Moreover, TAK-243 was effective against a high-risk myeloma model with deletion of p53 (Figure 5), and against primary samples and in vivo (Figures 6 and 7). Finally, combination regimens of TAK-243 with conventional and novel agents currently in use against myeloma showed potential for synergistic interactions (Table 2).
Three major proteasome activities have been described, including the chymotrypsin-like activity that cleaves after hydrophobic amino acids, the trypsin-like activity that cleaves after basic amino acids, and the post–glutamyl peptide (or caspase-like activity) that cleaves after acidic amino acids.46 This provides the proteasome with the ability to participate in turnover of virtually every cellular protein, and PIs, though very specific, therefore have a profound impact on protein homeostasis and cellular physiology.11 E1 inhibition could therefore at first be considered to have a more targeted impact because it would not influence turnover of proteins that are subject to proteasome-dependent but ubiquitin-independent proteolysis. Thus, from a purely proteostatic perspective, TAK-243 could be less toxic than PIs. Examples of proteins that undergo ubiquitin-independent proteolysis include Rpn4, thymidylate synthase, and ornithine decarboxylase,47 as well as, under some conditions, members of the Rb tumor-suppressor family.48 However, recent studies have found noncanonical pathways that mark proteins for proteasome-dependent turnover without the classical Lys48 polyubiquitination. These include polyubiquitination of other lysines, N-terminal residues, and internal threonine or serine residues, and monoubiquitination has recently been described as a signal for proteolysis.49 Notably, all of these processes depend on the availability of an ATP-activated ubiquitin moiety for target conjugation to occur, and therefore on UAE activity. Because UAE inhibition would deplete the pool of activated, thioester-linked ubiquitin, which is part of the cellular pool of free ubiquitin,50,51 this could disturb cellular homeostasis in a manner similar to that of PIs. Moreover, monoubiquitination is involved in other cellular processes, including endocytosis, intracellular localization, protein trafficking, histone modification, and DNA damage repair.52 Thus, UAE inhibition would be expected to also have a very broad impact on cellular biology, as in part evidenced by our findings on gene expression profiling (Table 1) and RPPA studies. Additional studies will be needed to better understand the different mechanisms of action downstream of these 2 types of agents, to better understand how UAE inhibition can overcome PI resistance, and to identify biomarkers of sensitivity to TAK-243.
One clear mechanism of action was through activation of ER stress and the UPR in our myeloma models, though a decrease in some stress proteins was seen in MM1.S cells at very high TAK-243 concentrations. Transcript and protein levels may differ because each provides only a snapshot of what is happening within the cell at that point in time. Also, one of the key adaptive effects of the ER stress response is to globally reduce new protein synthesis, so these reductions could be part of this effect. Moreover, ATF6 is subject to activation through its cleavage by site 1 and site 2 proteases during ER stress,53 whereas ATF4 has been described as a substrate for caspase-mediated cleavage.54 These data, and the finding of increased expression of MCL-1 and c-MYC in the presence of a caspase inhibitor (Figure 3B), suggest that such cleavage may be responsible for these changes. Several agents that have shown antimyeloma activity preclinically and clinically have done so through activation of the terminal, proapoptotic phase of the UPR, which has been dubbed an Achilles heel for myeloma cells.55 These include proteasome13-15 and HSP inhibitors,56 HIV protease inhibitors,57 and inhibitors of the AAA ATPase p97.58 Drugs that suppress the earlier, antiapoptotic activity of the UPR may also be active against myeloma, perhaps especially in combination with UPR inducers, to further enhance cell death. Examples may include dinaciclib59 and even doxorubicin,60 which may inhibit the IRE1α-XBP1 axis of the UPR, and may contribute through this mechanism to the efficacy of the bortezomib/pegylated liposomal doxorubicin regimen.61 Interestingly, our finding that UPR induction in myeloma cells may remain a reasonable approach to PI-resistant disease is encouraging, as patients with this history remain a clinical challenge and have a poor prognosis.3 Also, recent studies have suggested that PIs induce a prosurvival autophagic response through the UPR,62 and that sequestosome-1/p62-dependent autophagy may maintain proteostasis and determine susceptibility in myeloma cells.63 Therefore, suppression of autophagy may be an interesting approach to enhance myeloma sensitivity to proteasome inhibition,64 and the same could be true for UAE inhibition.
Blocking UAE activity with TAK-243 led to the accumulation of many short-lived proteins, such as p53 in cells with a WT TP53 gene. Interestingly, there was little to no difference in the sensitivity of 2 otherwise isogenic models of myeloma differing only in their TP53 status. These findings are encouraging considering that p53 activation induces a strong proapoptotic program, and other cell death pathways need therefore to be recruited to overcome this relative resistance if p53-mutant or deleted tumors are to be eliminated. Of note, Namba et al recently found that loss of p53 function activated IRE1α/XBP1, and that this pathway could serve as a target of chemoresistant tumors that expressed mutant p53.65 If true in myeloma, this could explain why our p53 knockout cells were just as responsive to UAE inhibition, and, indeed, showed greater induction of ATF6 than WT cells (Figure 5). Because p53 mutation remains a poor-risk feature in myeloma,45 these findings may indicate that TAK-243 could hold promise in this area.
Finally, we were able to show that TAK-243 combined well with other antimyeloma drugs, and was strongly synergistic with them, as indicated by very low CIs. Moreover, TAK-243 overcame resistance to conventional and novel drugs, including both bortezomib and lenalidomide. Together, these findings strongly support translation of this clinically relevant agent into trials targeting patients with relapsed and/or refractory myeloma, and possibly other hematologic malignancies as well.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
J.Z. acknowledges support from the Chinese Academy of Medical Sciences Initiative for Innovative Medicine (CAMS-2016-I2M-3-025). R.Z.O. acknowledges support received as the Florence Maude Thomas Cancer Research Professor, and support from the National Institutes of Health, National Cancer Institute (P50 CA142509, R01 CA184464, R01 CA194264, and U10 CA032102), the Leukemia & Lymphoma Society (SCOR-12206-17), the Adelson Medical Research Foundation, the Brock Family Myeloma Research Fund, and the Jean Clarke High-Risk Myeloma Research Fund. Core facilities that were key to these studies, including the Characterized Cell Line Core and the Flow Cytometry and Cellular Imaging Core, were supported by the MD Anderson Cancer Center Support Grant (National Cancer Institute P30 CA016672). Additional support came from the MD Anderson Cancer Center High-Risk Multiple Myeloma Moon Shot, the Yates Ortiz Myeloma Fund, and the Diane & John Grace Family Foundation.
Authorship
Contribution: J.Z. designed and performed most of the in vitro research; F.S., R.K.S., I.K., H.W., H.C.L., and R.J.J. performed additional laboratory studies, including with primary samples; Z.B. prepared manuscript drafts and provided helpful suggestions; A.B., M.H., N.C., S.S., J.Q.S., J.Y., V.S., and S.T. contributed in vivo modeling data; A.B. provided manuscript comments; Z.W. and R.E.D. performed gene expression profiling and provided helpful suggestions; and R.Z.O. supervised all the research completed herein and finalized the manuscript.
Conflict-of-interest disclosure: A.B., M.H., N.C., S.S., J.Q.S., J.Y., V.S., and S.T. are employees of Millennium Pharmaceuticals, Inc, a subsidiary of Takeda Pharmaceutical Company Limited (Cambridge, MA). R.Z.O. and H.C.L. have served on advisory boards for Millennium Pharmaceuticals, Inc/Takeda Pharmaceutical Company Limited. The remaining authors declare no competing financial interests.
Correspondence: Robert Z. Orlowski, Department of Lymphoma and Myeloma, The University of Texas MD Anderson Cancer Center, 1515 Holcombe Blvd, Unit 429, Houston, TX 77030-4009; e-mail: rorlowski@mdanderson.org.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal