

Abstract
Abstract 2801
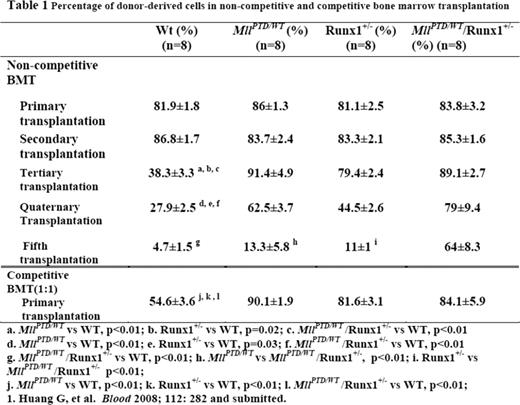
A relatively common mechanism through which Mixed-Lineage Leukemia (MLL) gene is disrupted in myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML) is a partial tandem duplication of its coding sequence (MLL-PTD), which duplicates the MLL N-terminal region and creates a larger in-frame oncogenic protein. We have previously shown that MLL interacts with and stabilizes the core binding factor (CBF) complex, a heterodimeric transcription factor complex composed of a DNA-binding subunit RUNX1 (CBFa) and a non-DNA-binding subunit CBFβ. MLL is required for CBF dependent regulation of the downstream target gene PU.11. In MDS and AML, RUNX1 is frequently disrupted by mutations, and the presence of MLL-PTD and/or RUNX1 mutations may negatively impact on clinical outcome. However, little is known how mutated MLL and RUNX1 contribute to MDS-associated ineffective hematopoiesis, clonal expansion and transformation into AML. Interestingly, MDS patients with either mutant RUNX1 or MLL-PTD tend to develop secondary AML harboring both RUNX1 and MLL mutations, suggesting a leukemogenic cooperation between the two genes through converging mechanisms. We hypothesized that the MLL-CBF nuclear signaling pathway maintains stem cell fitness, and the MDS-associated mutations co-opt this pathway leading to ineffective hematopoiesis, clonal expansion and transformation into AML. To address these questions, MLL-PTD+ and MLL-PTD(−) AML cell lines were compared. RUNX1 and CBFβ proteins were expressed at low levels in all 4 MLL-PTD+ AML cell lines (OCI-AML2, EOL, KOMP88, MUTZ11) compared with MLL-PTD(−) AML cell lines (OCI-AML5, Kasumi1, HL60, KG1a). In contrast to wild type MLL, which stabilized the CBF complex, MLL-PTD downregulated RUNX1 and CBFβ proteins in co-expression assays in 293T cells. This finding was further confirmed using the MllPTD/WT knock-in mouse model. In MllPTD/WT mice, the abnormal hematopoietic stem and progenitor cells (HSPCs) showed decreased expression levels of Runx1 (50%) and Cbfβ (30%) proteins and in turn PU.1 (30%) and its target gene, Mcl-1 (20%) compared with WT HSPCs. Consistent with this finding, HSPCs from MllPTD/WT mice had increased spontaneous apoptosis compared with WT HSPCs [(Lin−/cKit+/Sca-1+ (LSK), Annexin V positivity: 44% ±7.5% vs 25%±4.5%, p<0.05)]. At steady state, the bone marrow LSK and SLAM+LSK (CD150+/CD48−/LSK) populations were reduced by 50% (p<0.01) and 20% (p<0.01) respectively in MllPTD/WT mice compared with WT controls. Yet, when exposed to stress conditions (5-FU treatment), the HSPCs from MllPTD/WT mice exhibited rapid proliferation, reduced apoptosis and increased survival signaling with upregulated Bcl-xL compared with WT controls. Strikingly, HSPCs from MllPTD/WT mice exhibited enhanced expansion in serial bone marrow transplantation assays compared to those from WT controls (percentage of donor-derived cells: 91.4% ±4.9% vs 38.3% ±3.3% in tertiary, p<0.01; 62.5%±3.7% vs 27.9%±2.5% in quaternary transplantation, p<0.01). The enhanced expansion of MllPTD/WT HSPCs in recipient mice was similar to that of Runx1+/− HSPCs (percentage of donor-derived cells: 79.4%±2.4% in tertiary; 44.5%±2.6% in quaternary transplantation). In experiments using double heterozygous HSPC cells (MllPTD/WT/Runx1+/−), the enhanced expansion in recipient mice was even more pronounced (percentage of donor-derived cells: 89.1%±2.1% in tertiary; 79%±9.4% in quaternary; 64%±8.3% in 5th transplantation, p<0.01; table 1). These double heterozygous HSPC have even lower levels of Runx1 and Cbfb (20% and 10% respectively of the WT level). In a competitive bone marrow transplantation assay (at 1:1 ratio), the HSPCs of MllPTD/WT also show advantage in expansion compared with those from WT controls (90.1%±1.9% vs 54.6%±3.6% 16 weeks after transplantation, p<0.01; table 1). These data indicate that HSPCs of MllPTD/WT and Runx1+/− have similar enhanced expansion activity compared to wild type controls, while HSPCs of MllPTD/WT/Runx1+/− have even greater expansion activity compared to single mutant heterozygous HSPCs. This finding could partially explain the advantage of those HSPCs with both MLL-PTD and RUNX1 mutations in s-AML and de novo AML. Our findings provide a mechanistic model for development and maintenance of MLL-PTD- and RUNX1-related MDS and AML and provide novel targets for developing new therapies for these diseases.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal