Key Points
Matched sibling donor hematopoietic cell transplant and gene therapy both provided excellent overall survivals of 100% in ADA-SCID.
In patients without infections treated after 2000, no survival difference was found for hematopoietic cell transplant vs gene therapy.

Abstract
Adenosine deaminase (ADA) deficiency causes ∼13% of cases of severe combined immune deficiency (SCID). Treatments include enzyme replacement therapy (ERT), hematopoietic cell transplant (HCT), and gene therapy (GT). We evaluated 131 patients with ADA-SCID diagnosed between 1982 and 2017 who were enrolled in the Primary Immune Deficiency Treatment Consortium SCID studies. Baseline clinical, immunologic, genetic characteristics, and treatment outcomes were analyzed. First definitive cellular therapy (FDCT) included 56 receiving HCT without preceding ERT (HCT); 31 HCT preceded by ERT (ERT-HCT); and 33 GT preceded by ERT (ERT-GT). Five-year event-free survival (EFS, alive, no need for further ERT or cellular therapy) was 49.5% (HCT), 73% (ERT-HCT), and 75.3% (ERT-GT; P < .01). Overall survival (OS) at 5 years after FDCT was 72.5% (HCT), 79.6% (ERT-HCT), and 100% (ERT-GT; P = .01). Five-year OS was superior for patients undergoing HCT at <3.5 months of age (91.6% vs 68% if ≥3.5 months, P = .02). Active infection at the time of HCT (regardless of ERT) decreased 5-year EFS (33.1% vs 68.2%, P < .01) and OS (64.7% vs 82.3%, P = .02). Five-year EFS (90.5%) and OS (100%) were best for matched sibling and matched family donors (MSD/MFD). For patients treated after the year 2000 and without active infection at the time of FDCT, no difference in 5-year EFS or OS was found between HCT using a variety of transplant approaches and ERT-GT. This suggests alternative donor HCT may be considered when MSD/MFD HCT and GT are not available, particularly when newborn screening identifies patients with ADA-SCID soon after birth and before the onset of infections. This trial was registered at www.clinicaltrials.gov as #NCT01186913 and #NCT01346150.
Medscape Continuing Medical Education online
In support of improving patient care, this activity has been planned and implemented by Medscape, LLC and the American Society of Hematology. Medscape, LLC is jointly accredited by the Accreditation Council for Continuing Medical Education (ACCME), the Accreditation Council for Pharmacy Education (ACPE), and the American Nurses Credentialing Center (ANCC), to provide continuing education for the healthcare team.
Medscape, LLC designates this Journal-based CME activity for a maximum of 1.0 AMA PRA Category 1 Credit(s)™. Physicians should claim only the credit commensurate with the extent of their participation in the activity.
Successful completion of this CME activity, which includes participation in the evaluation component, enables the participant to earn up to 1.0 MOC points in the American Board of Internal Medicine’s (ABIM) Maintenance of Certification (MOC) program. Participants will earn MOC points equivalent to the amount of CME credits claimed for the activity. It is the CME activity provider’s responsibility to submit participant completion information to ACCME for the purpose of granting ABIM MOC credit.
All other clinicians completing this activity will be issued a certificate of participation. To participate in this journal CME activity: (1) review the learning objectives and author disclosures; (2) study the education content; (3) take the post-test with a 75% minimum passing score and complete the evaluation at http://www.medscape.org/journal/blood; and (4) view/print certificate. For CME questions, see page 795.
Disclosures
Laurie Barclay, MD, freelance writer and reviewer, Medscape, LLC, has disclosed the following relevant financial relationships: stock, stock options, or bonds: AbbVie Inc. (former).
Learning objectives
Upon completion of this activity, participants will:
- 1.
Describe baseline clinical, immunologic, and genetic characteristics in 131 patients with adenosine deaminase (ADA) deficiency causing severe combined immune deficiency (SCID) diagnosed between 1982 and 2017 who were enrolled in the Primary Immune Deficiency Treatment Consortium (PIDTC) SCID studies
- 2.
Determine treatment outcomes from first definitive cellular therapy (FDCT) in 131 patients with ADA-SCID diagnosed between 1982 and 2017 who were enrolled in the PIDTC SCID studies
- 3.
Identify clinical implications of baseline clinical, immunologic, and genetic characteristics and treatment outcomes from FDCT in 131 patients with ADA-SCID diagnosed between 1982 and 2017 who were enrolled in the PIDTC SCID studies
Release date: August 18, 2022; Expiration date: August 18, 2023
Introduction
Deficiency of the purine catabolic enzyme adenosine deaminase (ADA) is the cause for ∼13% of cases of severe combined immune deficiency (SCID).1,2 Absence of ADA activity results in adenosine and 2′-deoxyadenosine, along with their deoxyadenosine phosphorylated derivatives (dAXP), accumulating in multiple tissues.3-5 Excessive dAXP inhibits ribonucleotide reductase, blocks DNA synthesis and repair, and induces DNA breaks.6 ADA-deficient lymphocytes are particularly sensitive to these effects, leading to profound T-cell, B-cell, and natural killer (NK)-cell lymphopenia. Left untreated, the cellular and humoral immune deficiency results in life-threatening community acquired and opportunistic infections, as well as extra-immune complications, including pulmonary alveolar proteinosis,7-9 neurologic and neurocognitive deficits,10-12 sensorineural hearing loss,13,14 neutropenia and myeloid dysplasia,15-17 skeletal dysplasia,18,19 hepatic dysfunction,20,21 and malignant tumors.22-25
Treatment options for ADA-SCID include PEGylated ADA enzyme replacement therapy (ERT), allogeneic hematopoietic cell transplant (HCT), and ex vivo autologous gene therapy (GT). ERT enables systemic detoxification of adenine metabolites, promotes lymphopoiesis, and decreases opportunistic infections.26-31 Disadvantages of ERT include its high cost, dependence on once to twice weekly intramuscular injections, and frequent failure to achieve complete lymphocyte reconstitution.32 Complications in patients on long-term PEG-ADA include waning efficacy over time, breakthrough infections, autoimmunity, and malignancy.33-36 Although initial HCT approaches in the 1980s to 1990s were often not preceded by a period of ERT before HCT, recent consensus guidelines recommend ERT be initiated as soon as ADA-SCID is confirmed, bridging infants to definitive cellular therapy (HCT or GT).37 Concern has been raised, however, that ERT before HCT might negatively impact engraftment due to improvement in recipient immunity.
By comparison, HCT using a human leukocyte antigen (HLA) matched sibling or matched family donor (MSD/MFD), when available, is considered the optimal first definitive cellular therapy (FDCT) in ADA-SCID.37-39 In the largest, retrospective, multi-institution study of HCT for ADA-SCID between 1981 and 2009, 42 of 106 (41%) received MSD and 12 of 106 (13%) received MFD HCT, with overall survival (OS) of 86% and 83%, respectively.40 Most patients with MSD/MFD received unconditioned HCT, with 26 of 30 of these engrafting. This same study showed donor source and HLA-matching dramatically impacted outcomes, with recipients of matched unrelated donors (MUD), haploidentical donors (mismatched related donors [MMRD]), and mismatched unrelated donors (MMUD) having significantly worse OS of 67%, 43%, and 29%, respectively.40 These findings, along with promising results from recent GT trials,41 led many physicians to avoid alternative donor HCT and instead treat patients with ERT while awaiting GT. The impact of population-based SCID newborn screening (NBS), however, is important to consider in the current era of treatment.42 SCID NBS is now universally available in the United States and much of Canada and can modify the 2 most important predictors of successful HCT: absence of infection and HCT before 3.5 months of age.43-46 HCT outcomes, therefore, need re-examination in the contemporary era (after the year 2000) given NBS and general improvements in HCT over time, including refinements in HLA typing and supportive care.
Gammaretroviral vector transduced autologous CD34+ bone marrow cells were first reported to restore immunity in ADA-SCID by researchers from Milan, Italy.47-49 However, a case of T-cell leukemia in a patient with ADA-SCID 4 years following GT with the European Medicines Agency–approved gammaretrovirus product was reported in 2020,50 a reminder of the potential for genotoxic events with gammaretrovirus vectors.51-53 In recent years, GT has advanced to use lentiviral vectors, with clinical trials demonstrating excellent long-term gene correction of engrafted hematopoietic stem cells, immune reconstitution, event-free survival (EFS), and OS.41 GT requires low-intensity conditioning with single-agent busulfan.54 Importantly, recent GT trials have exclusively enrolled patients pretreated with ERT and without infection when starting the conditioning regimen, meaning the patient’s clinical status at FDCT was optimized. Despite the success of GT for ADA-SCID, worldwide availability is a concern. Clinical trials are currently limited and no commercial ADA-SCID GT exists in North America. Commercialized lentiviral gene-modified cell products, when available, may also be expensive.
Questions therefore remain regarding the optimal treatment approaches for ADA-SCID. (1) Does ERT before HCT impact survival? (2) How does immune reconstitution, EFS, and OS compare between HCT and GT? (3) How do HCT (including from alternative donors) and GT compare in the contemporary era, when infants are more likely to be diagnosed soon after birth and infection-free because of NBS?
The Primary Immune Deficiency Treatment Consortium (PIDTC) was established in 2009 to conduct multi-institutional observational studies of treatments for primary immunodeficiency diseases, including SCID.55 Despite the rarity of ADA-SCID (estimated to occur in 1 in 500 000 births),56 the prospective PIDTC 6901 and retrospective and cross-sectional PIDTC 6902 studies presented here encompass the largest cohort of patients with ADA-SCID reported to date, offering a comprehensive picture of the baseline clinical, molecular, and immunologic characteristics and outcomes following therapy for ADA deficiency.
Methods
Patients
All patients diagnosed with ADA-SCID between 1982 and 2017 at 27 North American PIDTC centers (supplemental Table 1 available on the Blood Web site) were enrolled on 1 of 2 PIDTC protocols. PIDTC 6901 (#NCT01186913), a prospective natural history study, enrolled newly diagnosed patients with SCID from 2010 forward, according to standard criteria.57 PIDTC 6902 (#NCT01346150), a retrospective and cross-sectional study, enrolled patients diagnosed with SCID between 1968 and 2012. Institutional review board approval for both protocols was obtained at each participating center. Participants and their guardians gave consent and assent for PIDTC 6901 and the cross-sectional component of PIDTC 6902 according to the Declaration of Helsinki. An institutional review board–approved waiver of informed consent was obtained for participants on the retrospective component of PIDTC 6902. Final eligibility for both protocols was determined by a PIDTC expert review panel.44
Inclusion as a case of ADA deficiency required at least 1 of the following: (1) deficient ADA enzyme activity (usually measured in erythrocytes or dried blood spots; occasionally in fibroblasts); (2) genetic confirmation of homozygous or compound heterozygous mutations known or predicted to result in ADA deficiency; or (3) an affected individual having a sibling (or other appropriate relative) with definitive ADA gene mutations. Management of ADA deficiency, including selection of ERT, HCT, GT, or combinations thereof, was determined by individual centers.
Data collection and definitions
Treating centers submitted patient data on PIDTC and Center for International Blood and Marrow Transplant Research case report forms (https://www.cibmtr.org/DataManagement/DataCollectionForms/Pages/index.aspx). Standard baseline, disease-specific, infectious, laboratory, and treatment-related data were collected at defined time periods, including at diagnosis, 100 days, 6 months, 1 to 2 years, 2 to 5 years, 6 to 10 years, and >10 years after initiation of ERT and each cellular therapy. A PIDTC ADA-SCID subcommittee queried treating centers regarding missing or unclear data to generate a final dataset for analysis.
Definitions were as follows: Trigger for diagnosis (NBS, family history, or infection), the single initial event leading to the eventual diagnosis of ADA-SCID; FDCT (first definitive cellular therapy) performed, either GT or HCT, regardless of the preceding use of ERT, subsequent initiation of ERT, or need for additional cellular therapy afterwards. Conditioning regimen intensity was divided into: (1) none; (2) immune suppression therapy (IST), which included serotherapy only or in combination with fludarabine and/or cyclophosphamide; (3) reduced intensity (RIC), which included regimens not meeting immune suppression therapy or myeloablative definitions (uniformly with lower dose busulfan); and (4) myeloablative (MAC), defined as any regimen using a cumulative busulfan dose ≥12 mg/kg. Serious infections included bacteremia (excluding coagulase-negative Staphylococcus or Bacillus), invasive fungal infections, Pneumocystis jiroveci, viral infections (eg, cytomegalovirus, adenovirus), Mycobacterium avium-intracellulaire, or vaccine-preventable infections (eg, Streptococcus pneumoniae, pertussis, mumps). EFS, days alive without restarting ERT or proceeding to a subsequent cellular therapy following FDCT; OS, days alive following FDCT.
Patients were grouped based upon whether they received: (1) ERT only with no cellular therapy; (2) HCT only (HCT); (3) HCT after ERT (ERT-HCT); or (4) GT after ERT (ERT-GT). GT with retroviral and lentiviral vectors was analyzed together for EFS and OS but separated for immune reconstitution analyses.
Statistics
Baseline characteristics were described for each group at the time of initial diagnosis and at FDCT. Comparisons between groups and for immune reconstitution analyses were performed using the Kruskal-Wallis test (quantitative values) and the χ2 or Fisher’s exact test (categorical variables). EFS and OS from the start of FDCT were estimated using the Kaplan-Meier method, with death (for EFS and OS) and restarting ERT or proceeding to a subsequent cellular therapy (for EFS) considered events. Data were censored at the date of last follow-up for surviving patients. Need for subsequent therapy after FDCT and acute and chronic graft-versus-host disease (GVHD) were analyzed using the cumulative incidence method, with death considered a competing event. Confidence intervals were calculated using a log-log transformation. Univariate comparisons between groups were performed using the log-rank test. Univariate analyses of risk factors associated with EFS, OS, and immune reconstitution in all patients receiving transplant (regardless of pretransplant ERT) were performed. No multivariate analyses were performed because of limited sample sizes. A subgroup analysis of EFS and OS, restricted to patients receiving FDCT after the year 2000 and without active infection at the time of FDCT, was performed to improve comparability between transplant and GT cohorts in the contemporary era. All analyses used SAS version 9.4.
Results
Patient characteristics
Baseline characteristics of the 131 patients with ADA-SCID studied (6901: n = 37; 6902: n = 94) are in Table 1. Most patients (n = 85, 64.8%) were diagnosed in the contemporary era, after the year 2000. Median age at diagnosis was 58 days (range: 0-4635 days). Most met criteria for typical SCID (85.5%), whereas 14.5% met criteria for leaky SCID.57 None had Omenn syndrome. Baseline lymphocyte proliferative response to phytohemagglutinin (PHA) was <10% of the lower limit of normal (LLN) in 70 of 83 (84.3% of cases with available data). No cases of maternal engraftment of lymphocytes were reported (0 of 19), although 112 of 131 (85.5%) had missing data. Infection was the most common reason to suspect ADA deficiency (57.5%), followed by a positive SCID NBS result (23.6%) and family history (18.9%). Weight and/or height < 5th percentile (68.1% and 53.1%, respectively) and need for respiratory support were common at diagnosis. Autoimmunity was uncommon (3.9%).
Evaluation of ADA enzyme activity and ADA gene mutations
One hundred eighteen participants (90%) had baseline diagnostic ADA enzyme and/or dAXP abnormalities (supplemental Materials; supplemental Figure 1). Thirty-six (27.5%) underwent ADA genotyping, which demonstrated 26 unique pathogenic mutations (supplemental Figure 2; supplemental Tables 2 and 3).
ERT alone increased lymphocyte numbers but to subnormal levels
To understand how lymphocyte numbers and function responded over time to ERT alone, lymphocyte subsets and PHA responses were analyzed by combining patients receiving ERT only (n = 9) with those receiving ERT before HCT or GT (n = 64), censored at the time of FDCT. Lymphocyte subsets increased in the first 6 months following initiation of ERT and then stabilized at lower-than-normal levels in subsequent years (Figure 1; supplemental Table 4). PHA responses were >30% LLN by 6 months after starting ERT in 8 of 11 patients with available data. Ten percent of patients discontinued IV immunoglobulin (IVIg) by 6 months on ERT only. To determine whether ERT before HCT or GT might elicit autoimmunity before FDCT (because of improved but dysregulated immunity), we compared the frequency of autoimmunity at the time of FDCT in the HCT group (n = 56) vs a combined ERT-HCT/ERT-GT group (n = 63, 1 with missing data). No difference was found (3.6% for HCT vs 7.9% for the combined ERT group; P = .45).
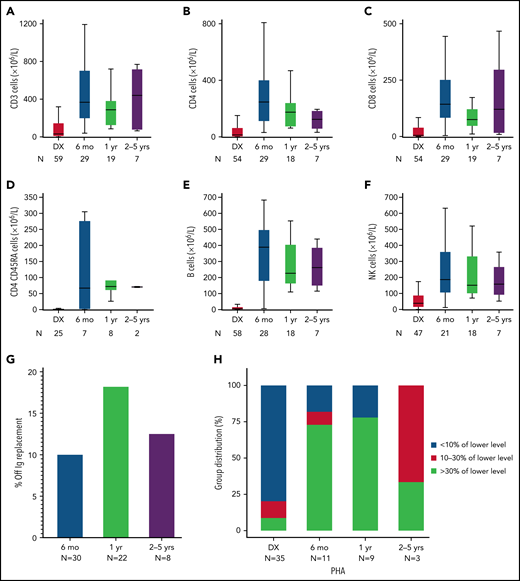
Immune reconstitution of T cells (CD3+, CD4+, CD8+, CD4+CD45RA+), B cells (CD19/20+), and NK cells (CD56+) while receiving ERT. Patients included those who received ERT only (n = 9) and patients who received ERT before first definitive cellular therapy, censored at the time of starting HCT or GT (n = 65). The x axis refers to the time after starting ERT. Numbers below each time point indicate the number of patients with evaluable data. (A-F) Absolute numbers of cells in each lymphocyte subset at ADA diagnosis (DX) and at different time points after initiation of ERT. By comparison, the 10th percentile of lymphocyte subsets in healthy 1- to 2-year-old children are as follows (all ×106/L): CD3+, 2100; CD4+, 1300; CD8+, 620; CD4+CD45RA+, 1000; CD19 B cells, 720; CD56 NK cells, 180.57 (G) Proportion of patients able to discontinue immunoglobulin replacement after ERT. (H) T-cell proliferative responses to phytohemagglutinin (PHA) improve for most but not all patients with ADA SCID after 6 months of ERT. Horizontal line, median; thick vertical bars, interquartile range; thin vertical lines, range.
Immune reconstitution of T cells (CD3+, CD4+, CD8+, CD4+CD45RA+), B cells (CD19/20+), and NK cells (CD56+) while receiving ERT. Patients included those who received ERT only (n = 9) and patients who received ERT before first definitive cellular therapy, censored at the time of starting HCT or GT (n = 65). The x axis refers to the time after starting ERT. Numbers below each time point indicate the number of patients with evaluable data. (A-F) Absolute numbers of cells in each lymphocyte subset at ADA diagnosis (DX) and at different time points after initiation of ERT. By comparison, the 10th percentile of lymphocyte subsets in healthy 1- to 2-year-old children are as follows (all ×106/L): CD3+, 2100; CD4+, 1300; CD8+, 620; CD4+CD45RA+, 1000; CD19 B cells, 720; CD56 NK cells, 180.57 (G) Proportion of patients able to discontinue immunoglobulin replacement after ERT. (H) T-cell proliferative responses to phytohemagglutinin (PHA) improve for most but not all patients with ADA SCID after 6 months of ERT. Horizontal line, median; thick vertical bars, interquartile range; thin vertical lines, range.
Initial treatment of ADA deficiency
Nine patients received ERT only. Two patients received cord blood derived GT (before the year 2000) and were excluded from further survival analyses, because this does not reflect contemporary GT practice. The remaining 120 patients who proceeded to FDCT included 56 (46.7%) who received HCT without initial ERT (HCT), 31 (25.8%) who received ERT before HCT (ERT-HCT), and 33 (27.5%) who received ERT before GT (ERT-GT; Table 2). Median duration of ERT before HCT or GT was 180 and 212 days, respectively (P = .51).
The 3 FDCT groups had clinically relevant differences at diagnosis and FDCT with potential to influence outcomes. Patients undergoing ERT-HCT and ERT-GT were more commonly diagnosed in the contemporary era, after the year 2000 (P < .001; Table 1). Decade of FDCT was also different, with 48.2% of HCT occurring after the year 2000 compared with 74.2% of ERT-HCT and 100% of ERT-GT (P < .001; Table 2). Patients undergoing HCT and ERT-HCT were more often diagnosed with ADA-SCID because of an infection or family history, whereas patients undergoing ERT-GT were more commonly identified by NBS (P = .021) and were therefore approximately 2 months younger at diagnosis (P = .04). By the time of ADA-SCID diagnosis, most patients undergoing HCT (67.9%) and ERT-HCT (75%) had 1 or more active or previously resolved infections. In contrast, 61.8% of patients undergoing ERT-GT had never experienced an infection (P = .002). Patients undergoing HCT and ERT-HCT in general were more ill compared with patients undergoing ERT-GT between diagnosis and FDCT, with higher proportions requiring supplemental oxygen (P = .005) or mechanical ventilation (P = .019). Higher proportions of patients receiving HCT and ERT-HCT also had active infections at the start of HCT (40.7% and 21.7%, respectively) compared with 0% of patients undergoing ERT-GT at the time of GT (P < .001). HCT patients were younger at time of FDCT (median, 131.5 days) compared with those receiving ERT-HCT (median, 361 days) or ERT-GT (median, 317 days; P < .001).
Differences in transplant approaches
Donor selection and conditioning differed between the HCT and ERT-HCT groups (Table 2). The HCT group more frequently received transplants from HLA-mismatched related donors (MMRD, haploidentical) (64.3%) compared with the ERT-HCT group (19.4% MMRD), who more frequently received transplants from an unrelated donor (URD) (61.3%) (P < .001). The HCT group also predominantly received unconditioned (69.6%) bone marrow grafts (89.3%) using older T cell depletion techniques, such as soybean lectin (57.1%). By comparison, the ERT-HCT group had higher proportions receiving cord blood (22.6%) or peripheral blood (16.1%) grafts, reduced intensity (26.7%) and myeloablative (46.7%) conditioning regimens, and serotherapy (51.6%) during conditioning. ERT-GT subjects uniformly received reduced intensity single-agent busulfan followed by autologous bone marrow CD34+ cells transduced by a retroviral (n = 12) or lentiviral (n = 21) vector.
EFS and OS and need for additional therapy after FDCT
Among the entire cohort, 5-year EFS was 49.5% for HCT, 73% for ERT-HCT, and 75.3% for ERT-GT (Figure 2A;,Table 3; P = .005). By univariate analysis, factors important for EFS in all patients receiving transplant (regardless of pretransplant ERT) included active infection at the time of transplant, donor source, and GVHD prophylaxis regimen. Age at transplant, ERT before transplant, trigger for diagnosis, and the conditioning regimen were not significant (Figures 2C,E,G; Table 3; supplemental Table 5).
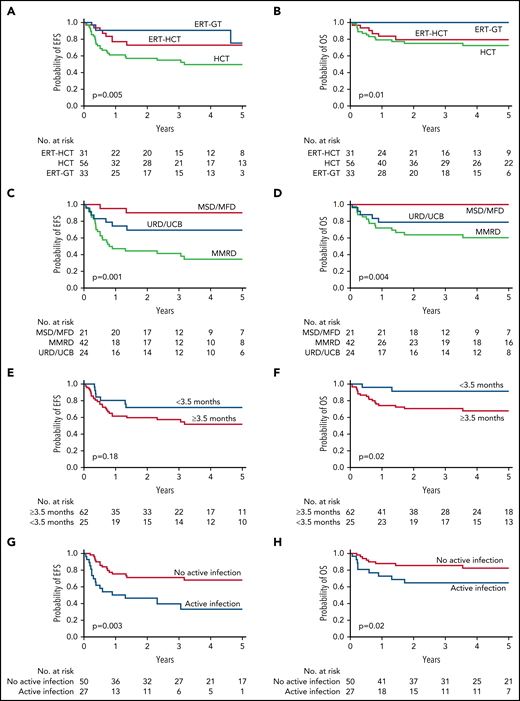
EFS and OS for ADA-SCID. (A) Five-year EFS by FDCT. (B) Five-year OS by FDCT. (C) Five-year EFS for all transplant patients (including those with and without a preceding period of ERT) by donor type. (D) Five-year OS for all transplant patients (including those with and without a preceding period of ERT) by donor type. (E) Five-year EFS for all transplant patients by age at the time of transplant. (F) Five-year OS for all transplant patients by age at the time of transplant. (G) Five-year EFS by the presence or absence of infection at the time of transplant. (H) Five-year OS by the presence or absence of active infection at the time of transplant.
EFS and OS for ADA-SCID. (A) Five-year EFS by FDCT. (B) Five-year OS by FDCT. (C) Five-year EFS for all transplant patients (including those with and without a preceding period of ERT) by donor type. (D) Five-year OS for all transplant patients (including those with and without a preceding period of ERT) by donor type. (E) Five-year EFS for all transplant patients by age at the time of transplant. (F) Five-year OS for all transplant patients by age at the time of transplant. (G) Five-year EFS by the presence or absence of infection at the time of transplant. (H) Five-year OS by the presence or absence of active infection at the time of transplant.
Five-year cumulative incidence of need for subsequent treatment after FDCT (starting ERT or undergoing a subsequent cellular therapy) was 30.1% for HCT (95% confidence interval [CI]: 17.7%-43.5%), 10.4% for ERT-HCT (95% CI: 2.5%-24.7%), and 24.7% for GT (95% CI: 3.3%-56.4%; P = .14; Figure 3). By univariate analysis, factors predictive of subsequent treatment after transplant (shown as 5-year estimated cumulative incidence) included donor source (MMRD 37.7% vs 9.1% for unrelated donor [URD]/umbilical cord blood [UCB] and 9.5% for MSD/MFD, P = .03) and conditioning regimen (none/IST 35.2% vs RIC/MAC 3.5%, P < .01; supplemental Table 6).
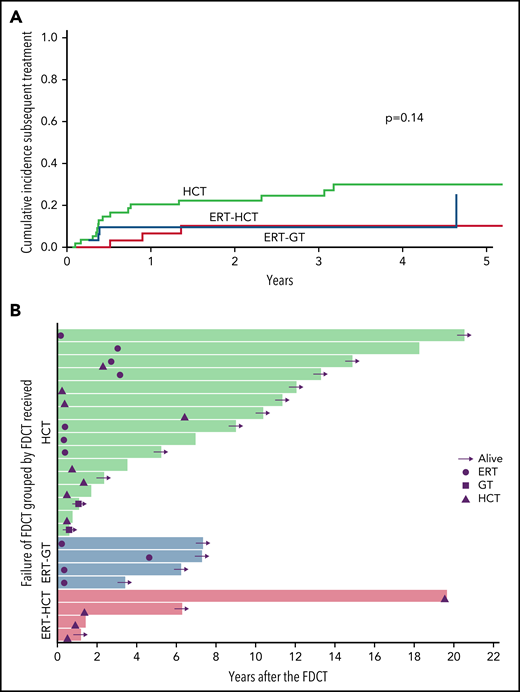
Failure of FDCT and subsequent treatments received after first definitive cellular therapy. (A) Cumulative incidence of patients receiving a second or greater therapy (ERT, GT, or HCT) after FDCT, according to FDCT received. (B) Swimmer plot showing second or greater therapy received after FDCT. Subsequent treatments are represented as circles (ERT), squares (GT), or triangles (HCT). Horizontal arrow means the patient is alive at last follow-up. No arrow means the patient died at that point in time after FDCT. Green bars represent patients receiving HCT as FDCT. Blue bars represent patients receiving ERT-GT as FDCT. All 4 patients receiving ERT-GT requiring subsequent therapy received retroviral vectors as FDCT (no patients receiving GT with lentiviral vectors required additional therapy), and ERT after GT as their second therapy. Red bars represent patients receiving ERT-HCT as FDCT.
Failure of FDCT and subsequent treatments received after first definitive cellular therapy. (A) Cumulative incidence of patients receiving a second or greater therapy (ERT, GT, or HCT) after FDCT, according to FDCT received. (B) Swimmer plot showing second or greater therapy received after FDCT. Subsequent treatments are represented as circles (ERT), squares (GT), or triangles (HCT). Horizontal arrow means the patient is alive at last follow-up. No arrow means the patient died at that point in time after FDCT. Green bars represent patients receiving HCT as FDCT. Blue bars represent patients receiving ERT-GT as FDCT. All 4 patients receiving ERT-GT requiring subsequent therapy received retroviral vectors as FDCT (no patients receiving GT with lentiviral vectors required additional therapy), and ERT after GT as their second therapy. Red bars represent patients receiving ERT-HCT as FDCT.
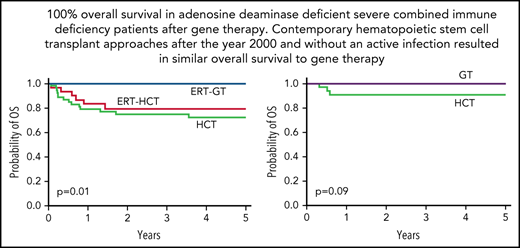
Five-year OS from FDCT was 72.5% for HCT, 79.6% for ERT-HCT, and 100% for ERT-GT (Figure 2B;,Table 3; P = .01). Sixteen of 56 (28.6%) patients undergoing HCT died compared with 7 of 31 (22.6%) patients undergoing ERT-HCT and 0 of 33 patients undergoing ERT-GT (supplemental Table 7). By univariate analysis, factors important for OS in all patients receiving transplant (regardless of pretransplant ERT) included age at transplant, active infection at transplant, donor source, GVHD prophylaxis regimen, and use of serotherapy (supplemental Table 8). OS was 100% for MSD/MFD, significantly better than URD/UCB (78.5%, P = .03) or MMRD (60.2%, P < .01; Figure 2D). Significantly better OS also occurred for patients transplanted at <3.5 vs ≥3.5 months of age (91.6% vs 67.9%, P = .02) and without vs with an active infection at the time of HCT (82.3% vs 64.7%, P = .02; Figure 2F,H). Factors not significantly impacting OS included use of ERT before HCT, trigger for diagnosis, and the conditioning regimen.
Immune reconstitution after FDCT
HCT, ERT-HCT, and ERT-GT using lentiviral (but not retroviral) vectors all showed similar improvements in CD3+, CD4+, and CD8+ T-cell counts out to 2 to 5 years following FDCT (Figure 4). Noteworthy is that immune reconstitution data were incomplete, particularly as patients became further away from FDCT (supplemental Table 9). Although no significant difference in CD3+, CD4+, and CD8+ T-cell counts existed between any cellular therapy by 6 months after FDCT, total T cells and their subsets were lower in the ERT-GT (retroviral) group compared with other cellular therapies at 1 and 2 to 5 years of follow-up. Notably, 4 of 12 patients undergoing ERT-GT (retroviral) restarted ERT after GT compared with 0 of 21 patients undergoing ERT-GT (lentiviral) (Figure 3B). Univariate analysis of factors associated with achieving >500 CD4+ T cells/µL by 2 to 5 years after transplant included donor type (highest with URD/UCB) and type of GVHD prophylaxis (highest with calcineurin inhibitors; supplemental Table 10), although patient numbers were small and dependent on patients still being alive with data reported. Naïve CD4+CD45RA+ T cells were also reported, although the number of evaluable patients was small. B-cell and NK-cell numbers were equivalent for the treatment groups at all time points after FDCT.
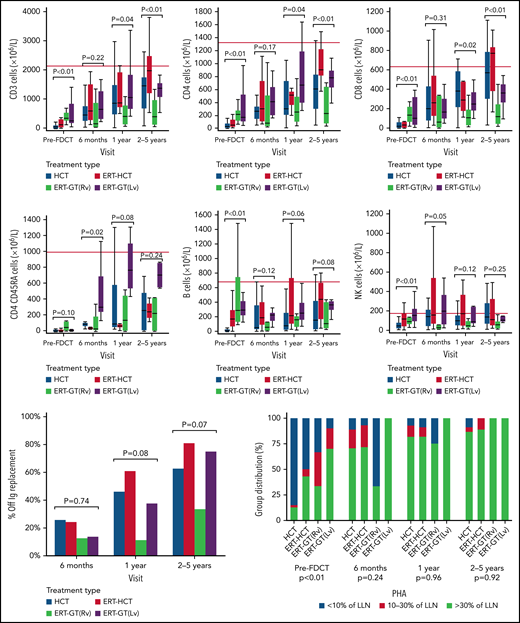
Immune reconstitution following first definitive cellular therapy. Dark horizontal line represents median. Thick vertical bars represent interquartile range. Thin vertical bars represent range. Red horizontal line represents the 10th percentile of lymphocyte subsets in healthy 1- to 2-year-old children.57 ERT-GT (Lv), enzyme replacement therapy followed by lentivirus gene therapy; ERT-GT (Rv), enzyme replacement therapy followed by retroviral gene therapy; FDCT, first definitive cellular therapy; Ig, immunoglobulin.
Immune reconstitution following first definitive cellular therapy. Dark horizontal line represents median. Thick vertical bars represent interquartile range. Thin vertical bars represent range. Red horizontal line represents the 10th percentile of lymphocyte subsets in healthy 1- to 2-year-old children.57 ERT-GT (Lv), enzyme replacement therapy followed by lentivirus gene therapy; ERT-GT (Rv), enzyme replacement therapy followed by retroviral gene therapy; FDCT, first definitive cellular therapy; Ig, immunoglobulin.
PHA responses were <10% LLN in most patients undergoing HCT before transplant. By comparison, PHA responses were significantly higher (P < .001) in patients undergoing ERT-HCT and ERT-GT before FDCT, associated with preceding ERT. After FDCT, differences in PHA responses were not significant between types of cellular therapy, with most patients achieving PHA responses > 30% LLN. Percentage of patients able to discontinue IVIg was similar in all treatment groups. Univariate analysis showed greater proportions of patients undergoing HCT and ERT-HCT without IVIg by 2 to 5 years after FDCT if they had received MSD/MFD or URD/UCB compared with MMRD (P = .038) or had received RIC/MAC conditioning vs none/IST (P = .02; supplemental Table 11). The number of patients with missing PHA and freedom from IVIg data, however, was high (64.7% and 48.3%, respectively).
After FDCT, the 5-year cumulative incidence of autoimmune cytopenias (HCT: 5.5% vs ERT-HCT: 3.3% vs ERT-GT: 0%; P = .43) and other autoimmune diseases (HCT: 2% vs ERT-HCT: 8.2% vs ERT-GT: 0%; P = .26) were not significantly different according to the type of FDCT received, with most autoimmune disease occurring in the first year after transplant.
Infections after FDCT
Seventy-three of 116 patients with complete data were reported to have 232 new infectious events after FDCT (range, 1-12; median, 3). Of these, 191 were considered serious, including non-tuberculous mycobacterial (n = 8), fungal/mold (n = 15) and vaccine-preventable (n = 34) infections. Serious infections occurred most frequently within the first year after FDCT and in recipients of HCT and ERT-HCT compared with ERT-GT, with a 1-year cumulative incidence of 35.3% (95% CI: 22.7%-48.1%), 31% (95% CI: 15.3%-48.3%), and 3.8% (95% CI: 0.3%-16.4%), respectively (P = .05). Infection was the cause of transplant-related mortality in 11 of 56 (19.6%) of recipients of HCT and 3 of 31 (9.7%) of recipients of ERT-HCT (supplemental Table 7).
Acute and chronic GVHD
Day 100 cumulative incidences of grade 2 to 4 acute GVHD for HCT and ERT-HCT were 15.2% (95% CI: 7%-26.2%) and 36% (95% CI: 14.9%-50.6%), respectively (P = .04), and for grade 3 to 4 acute GVHD were 5.7% (95% CI: 1.5%-14.2%) and 8% (95% CI: 1.3%-22.9%), respectively (P = .68). Two-year cumulative incidences of chronic GVHD for HCT and ERT-HCT were 3.8% (95% CI: 0.7%-11.6%) and 12.7% (95% CI: 3%-29.5%), respectively (P = .31).
No difference in survival between HCT and GT in contemporary patients with ADA-SCID without active infection at FDCT
With near universal NBS in the United States and Canada, and knowledge that survival outcomes for SCID are superior for patients receiving HCT before 3.5 months of age and without active infections,43-46 we sought to understand differences in survival between ADA-SCID patients receiving any HCT as FDCT after the year 2000 (reflecting contemporary HCT practice) and without an active infection at the time of transplant (n = 33) compared with patients with the same parameters receiving ERT-GT (n = 33; Table 4). Importantly, the HCT cohort comprised a wide variety of transplant approaches. The 5-year EFS and OS for HCT (75.3% and 90.9%, respectively) were not significantly different from ERT-GT (75.3% and 100%, respectively; Figure 5). Three patients undergoing HCT died, including 2 receiving unconditioned MMRD BMT with T-cell depletion using soybean lectin (1 with Clostridium difficile and rotavirus; 1 with pulmonary hypertension), and 1 receiving URD CD34+-selected peripheral blood stem cells following MAC (hepatic sinusoidal obstruction syndrome). Variation in HCT approaches and small patient numbers precluded analysis of individual factors predicting survival. Of the 33 HCT patients, 6 developed acute GVHD (grade II, n = 4; grade III, n = 2) and 2 developed limited chronic GVHD.
![Survival curves for ADA-SCID in a contemporary cohort receiving FDCT after the year 2000 and without active infection at the time of definitive cellular therapy. The HCT group (n = 33) comprised a wide variety of transplant approaches (Table 4), including patients who did (n = 15) and did not (n = 18) receive ERT before HCT; and a wide variety of donor types (MSD [n = 9], MFD [n = 3], unrelated donors [n = 13], and MMRD [n = 8]), graft sources (bone marrow [n = 21], cord blood [n = 8], and peripheral blood [n = 4)], and conditioning regimens (none [n = 15], RIC [n = 6], and MAC [n = 11]). The contemporary HCT group was compared against patients receiving ERT-GT (n = 33). (A) Five-year EFS for the HCT group was 75.3% (95% CI: 56.5%-86.8%) vs 75.3% (95% CI: 34.3%-92.7%) for the GT group (P = .29, log-rank test). (B) Five-year OS for the HCT group was 90.9% (95% CI: 74.4%-97%) vs 100% (95% CI: 100%-100%) for the GT group (P = .09, log-rank test).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/7/10.1182_blood.2022016196/4/m_bloodbld2022016196f5.png?Expires=1768017100&Signature=Sf~sL~hLW1Bm5eUj9TYroJAsS5rzLoblSID6CLb~gJC0iGFnuzGb191E2oa4zywrpCZJuJ4v~zcRVYzfcXdaiGmIVkhnTOKDWEXkiLrTb0RNrt2Of1Irz-1C0kqM6wOXoj6jX2Vn2W7T-ybon4Pf~8kM-B6Nzo7QfPvUgfL1leRULT72sPOPwm7SgXCok457YRRVIpbXx2p5HfGZussO06e0hU3CBK3weHovgDZicy33HIPBROb2CSwd3~bZskcftKcA1QdmoAEUI8bRlE5GU1yAiQ8Qrrc~I0LCws5TK4MVI6Jf40bjoK8rfvr~uGWK8wbxGMn1LGyMEpWz-yyL1w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Survival curves for ADA-SCID in a contemporary cohort receiving FDCT after the year 2000 and without active infection at the time of definitive cellular therapy. The HCT group (n = 33) comprised a wide variety of transplant approaches (Table 4), including patients who did (n = 15) and did not (n = 18) receive ERT before HCT; and a wide variety of donor types (MSD [n = 9], MFD [n = 3], unrelated donors [n = 13], and MMRD [n = 8]), graft sources (bone marrow [n = 21], cord blood [n = 8], and peripheral blood [n = 4)], and conditioning regimens (none [n = 15], RIC [n = 6], and MAC [n = 11]). The contemporary HCT group was compared against patients receiving ERT-GT (n = 33). (A) Five-year EFS for the HCT group was 75.3% (95% CI: 56.5%-86.8%) vs 75.3% (95% CI: 34.3%-92.7%) for the GT group (P = .29, log-rank test). (B) Five-year OS for the HCT group was 90.9% (95% CI: 74.4%-97%) vs 100% (95% CI: 100%-100%) for the GT group (P = .09, log-rank test).
Survival curves for ADA-SCID in a contemporary cohort receiving FDCT after the year 2000 and without active infection at the time of definitive cellular therapy. The HCT group (n = 33) comprised a wide variety of transplant approaches (Table 4), including patients who did (n = 15) and did not (n = 18) receive ERT before HCT; and a wide variety of donor types (MSD [n = 9], MFD [n = 3], unrelated donors [n = 13], and MMRD [n = 8]), graft sources (bone marrow [n = 21], cord blood [n = 8], and peripheral blood [n = 4)], and conditioning regimens (none [n = 15], RIC [n = 6], and MAC [n = 11]). The contemporary HCT group was compared against patients receiving ERT-GT (n = 33). (A) Five-year EFS for the HCT group was 75.3% (95% CI: 56.5%-86.8%) vs 75.3% (95% CI: 34.3%-92.7%) for the GT group (P = .29, log-rank test). (B) Five-year OS for the HCT group was 90.9% (95% CI: 74.4%-97%) vs 100% (95% CI: 100%-100%) for the GT group (P = .09, log-rank test).
Discussion
ADA-SCID, a systemic metabolic disorder with nonlymphoid manifestations and potential to rescue lymphocyte development with exogenous enzyme, is distinct from other forms of SCID. The 131 patients with ADA-SCID enrolled in the PIDTC 6901/6902 studies and reported here provide a detailed view of this rare inborn error of immunity and its treatments. Previous publications from the PIDTC included only limited numbers of patients with ADA-SCID receiving HCT.43,44,58,59 In contrast, this genotype-specific analysis is more comprehensive, encompassing all treatment approaches. Our data confirm that most cases of ADA deficiency present as typical SCID because of severe T-cell, B-cell, and NK-cell deficiency, although 14.5% presented with a “leaky” SCID phenotype.57 Correlations have previously been established between ADA genotype, residual ADA enzyme activity, and clinical phenotype.60,61 We wanted to understand whether certain ADA mutations also correlated with treatment outcomes. Genotype data, however, were only available for 36 of 131 cases, which was insufficient for analysis. No cases of maternal engraftment were found, although 85.5% of cases had no data, limiting any conclusion concerning maternal engraftment in ADA-SCID. Ongoing PIDTC efforts are emphasizing the importance of testing for maternal engraftment in all patients with a suspected or confirmed SCID diagnosis, given that the results may be informative in classifying typical vs leaky SCID in updated PIDTC SCID diagnostic criteria (unpublished, revised PIDTC SCID criteria, 2022) while informing further about the immunobiology of SCID disorders.
Because patients in our cohort were diagnosed as early as 1982, this analysis provides historical perspective on the treatment of ADA-SCID over 4 decades. Earliest HCT methodology involved unconditioned MSD/MFD and MMRD using T cell–depleted bone marrow, without ERT for ADA-SCID.45,62-65 This approach saved the lives of many infants with ADA-SCID, a high proportion of whom were suffering from serious infections.66 Our data continue to support excellent outcomes of MSD/MFD HCT for ADA-SCID, with 5-year EFS and OS of 90.5% and 100%, respectively. Thus, when available, MSD/MFD HCT remains a preferred FDCT for ADA-SCID,37 with transplant occurring urgently following confirmation of the diagnosis and before the onset of infections. In the current era of SCID NBS, this can be accomplished soon after birth and ideally before 3.5 months of age.
Other HCT approaches have been attempted in ADA-SCID. In our cohort, MMRD had the lowest 5-year EFS and OS at 34.6% and 60.2%, respectively, significantly inferior to MSD/MFD (EFS and OS) and URD/UCB (EFS only) (Figure 2). However, MMRD HCT was often performed in earlier treatment eras, using unconditioned parental haploidentical bone marrow (T cell–depleted by older techniques, such as soybean lectin) and in patients with active infections (a poor prognostic factor), with many requiring subsequent cellular therapies (HCT or GT) after FDCT. Our data do not allow firm conclusions about the role of MMRD HCT in the contemporary era (since 2000) in uninfected infants, because only 8 patients received MMRD HCT in this subset analysis (6 are alive). Furthermore, none of these patients underwent MMRD HCT with posttransplant cyclophosphamide or depletion of αβT-cell receptor T cells, advocated by some to reduce GVHD.67,68 Moreover, our data were insufficient to determine the role of haploidentical HCT for ADA-SCID now that infants are largely diagnosed by NBS and brought to transplant without infections. When no other curative options exist, MMRD HCT vs indefinite ERT must be considered on a case-by-case basis.
We demonstrated that T-cell, B-cell, and NK-cell numbers increased in the first 6 months after ERT initiation, followed by a plateauing of lymphocyte subsets within 1 year of continuous ERT, to levels significantly below those of healthy 1- to 2-year-old children. Furthermore, ERT before transplant (when considering all transplant patients) had no impact on 5-year EFS or OS by univariate analysis, and the cumulative incidence of need for a subsequent therapy after FDCT (whether restarting ERT or a second or greater HCT) among HCT, ERT-HCT, and ERT-GT groups was not different, suggesting ERT before FDCT did not appreciably increase graft rejection. The high cost of ERT and less than normal immune reconstitution with prolonged ERT must be weighed against the possibility of avoiding serious infections and metabolic toxicity (including pulmonary alveolar proteinosis)4 if ERT is to be considered as a bridge therapy while awaiting transplant. Data required to determine whether and, if so, when to discontinue ERT before transplant are not available. These remain important questions for the future.
Outcomes after ERT-GT (retroviral and lentiviral) were excellent, with 5-year EFS and OS of 75.3% and 100%, respectively. Although immune reconstitution of CD3+, CD4+, and CD8+ T cells was similar between patients treated with lentiviral ERT-GT, HCT, and ERT-HCT, T-cell recovery following retroviral ERT-GT was inferior 1 year after FDCT, with 4 of 12 patients receiving retroviral ERT-GT needing to restart ERT. As reported in a prior series,40 the absolute numbers of lymphocytes achieved in the patients with ADA SCID were lower than normal; the basis for this is unknown but may reflect early damage to lymphoid niches from ADA deficiency before therapy. Unfortunately, GT for ADA-SCID is currently limited in North America. Although we are aware of some patients with ADA-SCID without MSD/MFDs receiving long-term ERT while awaiting GT, our data suggest that pursuing alternative donor HCT (particularly HLA-matched URD/UCB) for young infants without infection should be considered an alternative, given no difference in EFS and OS between HCT and GT since the year 2000. A recent single-center study suggested similar EFS and OS between MUD and MSD/MFD, although numbers were small.69
The next PIDTC 6907 SCID study is opening at 47 centers in North America and will help to answer remaining questions about ADA-SCID, including outcomes of cellular therapy in the era of NBS, the optimal time to discontinue ERT before FDCT, the role of newer approaches to haploidentical HCT, and long-term neurodevelopmental outcomes.
Acknowledgments
The authors thank PIDTC Project Managers Sharon Kidd, Elizabeth Dunn, and Kiana Soriano for their dedicated work to advance the research goals of the consortium; all study coordinators at PIDTC sites for collection of clinical data from medical records; the clinical teams who provided medical care for patients; and all the patients and families who have made this work possible.
This work was supported by the Division of Allergy, Immunology and Transplantation, National Institute of Allergy and Infectious Diseases (NIAID), the Office of Rare Diseases Research (ORDR), National Center for Advancing Translational Sciences (NCATS), National Institutes of Health (NIH) (grant U54AI082973, MPI: J.M.P., C.C.D., E.H.; grants U54NS064808 and U01TR001263). The PIDTC is a part of the Rare Diseases Clinical Research Network of ORDR, NCATS. The collaborative work of the PIDTC with the Pediatric Transplantation and Cellular Therapy Consortium is supported by the U54 grants listed, along with support of the PBMTC Operations Center by the St. Baldrick’s Foundation and grant U10HL069254. Collaborative work of the PIDTC with the Center for International Blood and Marrow Transplant Research is supported by grant U24CA076518, grant U01HL069294, contracts HHSH250201200016C and HHSH234200637015C with the Health Resources and Services Administration, and grants N00014-13-1-0039 and N00014-14-1-0028 from the Office of Naval Research. L.D.N. is supported by the Division of Intramural Research, NIAID, NIH (grant 1 ZIA AI001222-02, PI: L.D.N). S.-Y.P. is supported by funding from the Intramural Research Program, NIH, National Cancer Institute, Center for Cancer Research.
The content and opinions expressed are solely the responsibility of the authors and do not represent the official policy or position of the NIAID, ORDR, NCATS, NIH, HRSA, or any other agency of the US Government.
The sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Authorship
Contribution: G.D.E.C. is part of the ADA-SCID PIDTC working group, contributed patients, designed the study, performed data analysis, and was primarily responsible for writing of the manuscript; D.B.K. is the senior leader of the ADA-SCID PIDTC working group, contributed patients, designed the study, performed data analysis, and was primarily responsible for writing of the manuscript; S.E.P., R.H.B., C.Y.K., L.M.G., A.Y., E.H., and J.M.P. are members of the ADA-SCID PIDTC working group, contributed patients, designed the study, performed data analysis, and co-wrote and edited the manuscript; B.R.L. and X.L. provided all biostatistical support; M.S.H. provided data and analysis regarding ADA enzyme activity and ADA gene mutations; P.G.A. provided the figure for the location of specific ADA gene mutations; and M.J.D., R.E.P., C.L.E., T.B.M., R.J.O, S.-Y.P., M.K., N.K., L.R.F.S., C.M., L.M.B., A.P., M.S.T., D.C., J.H., D.C.S., A.R., J.J.B., S. Chandra, S. Chandrakasan, A.P.G., L.M., T.C.Q., E.H.C., B.J.D.S., K.D., H.E., F.D.G., J.R., A.J.S., M.T.V.L., M.A.P., L.D.N., M.J.C., J.W.L., T.R.T., and C.C.D. provided patients, helped with data cleaning, and edited the manuscript.
Conflict-of-interest disclosure: G.D.E.C. has received consultancy fees from Miltenyi Biotech. D.B.K. is an inventor for the UC Regents on a lentiviral vector for gene therapy of ADA SCID and is a member of the DSMB for Revcovi PEG-ADA (Chiesi, USA). S.E.P. has received clinical trial support from Atara Biotherapeutics, Jasper Pharmaceuticals, and AlloVir; is an inventor of IP licensed to Atara Biotherapeutics by Memorial Sloan-Kettering (all rights assigned to MSK); and has been part of advisory boards for ADMA and Neovii. J.M.P. has received royalties from Up-To-Date, and her spouse is employed by and owns stock in Invitae (a DNA sequencing company). E.H. has received consultancy fees from Chiesi, USA (producers of Revcovi PEG-ADA), has received adboard meeting fees for CSL-Behring and Takeda and DSMB fees for Jasper Therapeutics and Rocket Pharmaceutical, and is involved in Immugenia, a biotech company. M.J.D. has received institutional research support from Chiesi, USA. C.C.D. is a member of the DSMB for Revcovi PEG-ADA (Chiesi, USA) and has received consultancy fees from Orchard Therapeutics. J.J.B. has served on the advisory board for Sobi and Horizon Therapeutics. M.S.H has received grant support for his laboratory from Chiesi, USA (producers of Revcovi PEG-ADA) and Orchard Therapeutics. M.A.P. has served on the advisory boards of Novartis, Medexus, Equillium, and Mesoblast; has received clinical study support from Adaptive and Miltenyi Biotech; and has received financial support for educational lectures for Miltenyi Biotech and Novartis. M.S.T. has been a consultant for the Infectious Disease Research Institute (nonprofit). L.R.F.S. has received consultancy fees from Enzyvant, CSL Behring, Takeda, ADMA, and Grifols. B.J.D.S. has received consultancy fees from Sobi and Orchard. J.H. has received clinical trial support from Regeneron, an investigator-initiated grant, and participated on an advisory board for CSL Behring; has received royalties from Up-To-Date; and has participated on an advisory board for ADMA Biologics. J.W.L. has been a speaker and consultant for Horizon Therapeutics, speaker for Sobi, consultant for ADMA Biologics, and is an employee and shareholder of Bluebird Bio. A.P. has participated in advisory boards for Orchard, Horizon, and Enzyvant and serves on the DSMB for ExCellThera. L.M.B. has received clinical trial support through the Fred Hutchinson Cancer Research Center by Medac GmbH (including supply of treosulfan), is a member of the DSMB for a clinical trial with Jasper Therapeutics, and has served on an advisory board for Horizon Therapeutics USA. E.H.C. has received royalties for participation in advisory boards from Pfizer. M.J.C. has received royalties from Up-To-Date, is on the Scientific Review Board of Homology Medicines with equity interest, and is on the DSMB for clinical trials with Bluebird Bio, Rocket Pharmaceuticals, and Chiesi, USA. The remaining authors declare no competing financial interests.
Correspondence: Geoffrey D. E. Cuvelier, Division of Pediatric Hematology-Oncology-BMT, 2021a-675 McDermot Ave, CancerCare Manitoba, Winnipeg, MB R3E 0V9, Canada; e-mail: gcuvelier@cancercare.mb.ca.
Requests for data sharing may be submitted to Reem Mustafa (rmustafa@kumc.edu).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.