Abstract
Castleman disease (CD) describes a group of at least 4 disorders that share a spectrum of characteristic histopathological features but have a wide range of etiologies, presentations, treatments, and outcomes. CD includes unicentric CD (UCD) and multicentric CD (MCD), the latter of which is divided into idiopathic MCD (iMCD), human herpes virus-8 (HHV8)-associated MCD (HHV8-MCD), and polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, skin changes (POEMS)-associated MCD (POEMS-MCD). iMCD can be further subclassified into iMCD–thrombocytopenia, ascites, reticulin fibrosis, renal dysfunction, organomegaly (iMCD-TAFRO) or iMCD–not otherwise specified (iMCD-NOS). Advances in diagnosis, classification, pathogenesis, and therapy are substantial since the original description of UCD by Benjamin Castleman in 1954. The advent of effective retroviral therapy and use of rituximab in HHV8-MCD have improved outcomes in HHV8-MCD. Anti–interleukin-6–directed therapies are highly effective in many iMCD patients, but additional therapies are required for refractory cases. Much of the recent progress has been coordinated by the Castleman Disease Collaborative Network (CDCN), and further progress will be made by continued engagement of physicians, scientists, and patients. Progress can also be facilitated by encouraging patients to self-enroll in the CDCN’s ACCELERATE natural history registry (#NCT02817997; www.CDCN.org/ACCELERATE).
Introduction
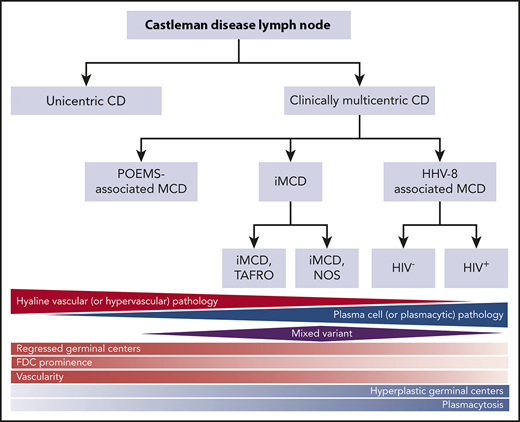
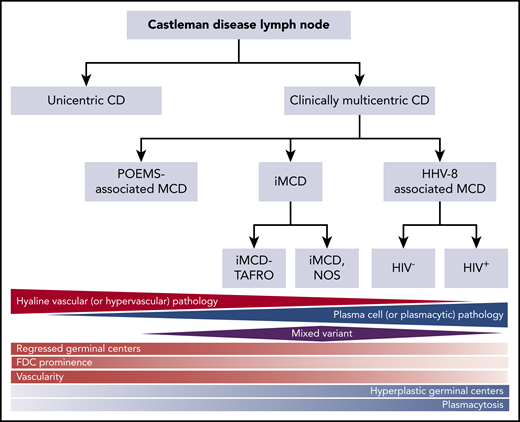
Castleman disease (CD) describes a group of at least 4 disorders that share a spectrum of characteristic histopathological features but have a wide range of etiologies, presentations, treatments, and outcomes. CD was first described in the 1950s by Benjamin Castleman as localized mediastinal lymph node enlargement characterized by increased numbers of lymphoid follicles with germinal center involution and marked capillary proliferation, including follicular and interfollicular endothelial hyperplasia.1 In 1969, Flendrig described the plasma cell (PC), the hyalinized, and the “intermediate” (or mixed) histopathological variants.2,3 Further descriptions over the years provided insight into clinicopathologic associations.3,4 By the mid-1980s, CD was divided into unicentric CD (UCD), which involved a single enlarged lymph node or region of lymph nodes, and multicentric CD (MCD), which involved multiple lymph node stations.5,6 Investigators also noted an association between HIV and MCD.7,8 Co-occurrence with and overlap between the PC neoplasm polyneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder, skin changes (POEMS) syndrome (also known as Takatsuki or Crow-Fukase) and MCD was also noted in the 1980s and 1990s; later, the monoclonal PCs causing POEMS were proposed to be causing the MCD in these cases. Human herpes virus-8 (HHV8) was identified as the etiological driver of all HIV+ and some HIV− MCD cases in the 1990s. In the 2010s, Takai et al recognized a severe form of HHV8− or idiopathic MCD (iMCD) in which patients had a homogeneous constellation of abnormal laboratory tests and clinical features that he called thrombocytopenia, ascites, reticulin fibrosis, renal dysfunction, organomegaly (TAFRO) syndrome.9,10 Recently, the Castleman Disease Collaborative Network (CDCN) proposed a classification system retaining the UCD vs MCD nomenclature, but further dividing MCD by etiological driver (HHV8-associated MCD [HHV8-MCD]; POEMS-associated MCD [POEMS-MCD]; iMCD) and within iMCD by phenotype, iMCD-TAFRO, and iMCD–not otherwise specified (iMCD-NOS) (Figure 1).11
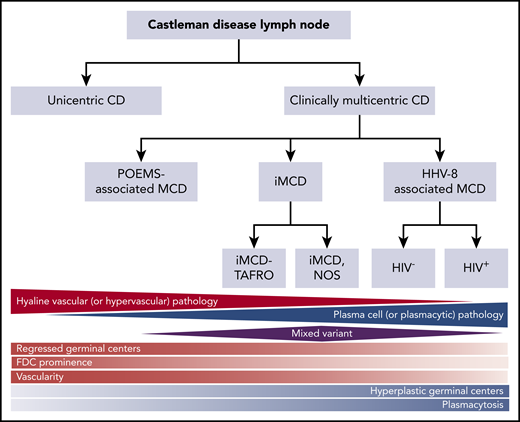
Classification of CD. Triangles and diamond represent the relative frequency that each of these pathologies occurs within the different subtypes of CD. The relative red and blue shading reflects the extent to which each of the pathology types contains either hypervascular or plasmacytic pathology. FDC, follicular dendritic cell. Adapted from Fajgenbaum et al.11
Classification of CD. Triangles and diamond represent the relative frequency that each of these pathologies occurs within the different subtypes of CD. The relative red and blue shading reflects the extent to which each of the pathology types contains either hypervascular or plasmacytic pathology. FDC, follicular dendritic cell. Adapted from Fajgenbaum et al.11
Epidemiology
The epidemiology of CD is poorly studied. Male subjects are slightly more often affected with MCD than female subjects, but for UCD there is no gender preference. The average age of diagnosis for UCD patients is typically younger (fourth decade) than for MCD patients (sixth decade), but patients of all ages, including young children, can be diagnosed with any form of CD.5,12-14
No known risk factors exist for UCD, POEMS-MCD, or iMCD. Immunocompromise is the primary risk factor for HHV8-MCD, and HIV is one of the most common chronic immunocompromised states underlying HHV8-MCD.15 Virtually all HIV+ MCD patients have HHV8-MCD; whereas, in the HIV− MCD population, HHV8-MCD accounts for 2% to 50% of MCD cases, though this latter estimate is highly dependent on how endemic HHV8 is in the overall population.16-20 Additional risk factors for HHV8-MCD include country of origin,21 consanguineous parentage, thymoma, chronic viral hepatitis, organ transplant, and men having sex with men.17
The estimated annual incidence of UCD and MCD in the United States is 4300 to 5200, but other studies have estimated a lower incidence.22-24 Unlike Kaposi sarcoma, whose incidence has reduced in the combination antiretroviral therapy era, recognition of HIV+ HHV8-MCD has increased. In a large prospective database of HIV+ individuals, 24 with HHV8-MCD were identified for an overall incidence of 5.3 per 10 000 patient-years (95% confidence interval, 2.4, 6.4).25
Pathogenesis
Data suggest that UCD is more likely a clonal neoplastic process,26 and the most likely cell of origin is stromal, specifically the follicular dendritic cell.7,8,26 A recent study using next-generation sequencing of UCD lymph node tissue revealed somatic platelet-derived growth factor receptor β mutations in nearly 20% of cases27 ; the mutations were localized to CD45− cells, likely representing stromal cells. In vitro experiments confirmed that the mutation is a gain of function that confers proliferation and survival advantages.
Uncontrolled HHV8 infection is the etiological driver in HHV8-MCD.28 In immunocompromised individuals, HHV8 can replicate in lymph node plasmablasts and transcribe the viral homolog of interleukin-6 (IL-6; vIL-6) that drives symptoms, signs, and lymph node pathology along with a cascade of other cytokines including human IL-6.15 It has been postulated that HHV8 may be able to cause immunoglobulin M–positive (IgM+) naive B cells to differentiate into plasmablasts without undergoing the germinal center reaction.29 Human IL-6 expression can be localized to germinal center follicular dendritic cells and PCs30,31 ; whereas, vIL-6 is localized to the mantle zone and interfollicular regions19,31,32 (see excellent recent review).33
The etiology and pathogenesis of HHV-8−/iMCD is less well understood than HHV8-MCD or POEMS-MCD. IL-6 is a critical disease driver in some patients as demonstrated by abrogation of iMCD signs and symptoms with IL-6 and IL-6 receptor antibodies34,35 and recapitulation of the iMCD phenotype with overexpression of IL-6 in mice.36 Vascular endothelial growth factor (VEGF) is also elevated in iMCD patients, but less so than in POEMS.37 Although IL-6 is the driver of iMCD pathogenesis in some patients, IL-6 is not universally elevated in iMCD, and approximately one-half of iMCD patients do not respond to IL-6 inhibition. Alternative cytokines and signaling pathways are likely involved in these cases. Recently, a key role for activated T cells and mammalian target of rapamycin pathway activation has been observed, which has led to the use of an mammalian target of rapamycin inhibitor with early success.38
The cause of the increased IL-6 and other cytokines in iMCD is unknown. Speculation regarding whether iMCD is an autoimmune, infectious, or clonal disease abounds, but data are limited.39 The hypothesis that iMCD is driven by infection with a virus other than HHV8 is becoming less credible.40 Twenty-five patients with CD were studied using an RNA-hybrid, deep sequencing, and bioinformatics virome capture sequencing platform to search for vertebrate viruses.40 None of the 11 iMCD or the 12 UCD patients had HHV8 identified in their frozen lymph nodes specimens; only the 2 known HHV8+ MCD patients were positive for HHV8. No novel viruses were discovered, and there were no clear associations between UCD or iMCD and any known virus. To further test the pathogen hypothesis, CD samples are being analyzed using an orthogonal method that detects nucleotide sequences from all known viruses, bacteria, fungi, and parasites.39 Given the apparent overlap in symptoms, signs and lymph node pathology with autoimmune conditions, the notion of an autoimmune etiology for iMCD has some appeal although more data are needed. Autoantibodies are observed in approximately one-third of iMCD patients,41 and a polymorphism in IL-6R was increased among iMCD patients compared with healthy controls.42 Finally, the hypothesis of neoplastic cells driving iMCD is appealing given the increased risk of malignancy as compared with age-matched controls and the clinicopathologic overlap with malignancies like Hodgkin lymphoma and myelofibrosis.41,43 Further studies are needed.
Monoclonal PCs are the likely etiological drivers of POEMS-MCD. Exactly how the cellular and cytokine profiles between POEMS-MCD and classic POEMS syndrome differ is unknown, but excessive VEGF and IL-12 production due to somatic mutations in PCs are the established drivers in classic POEMS syndrome.44 A comprehensive genetic analysis on the PCs of 20 patients with classic POEMS syndrome revealed 20 mutations in 7 recurrently mutated genes: KLHL6, LTB, EHD1, EML4, HEPHLI, HIPK1, and PCDH10.45 Surprisingly, VEGFA expression was not increased in the PCs of POEMS patients. In contrast, Wang et al demonstrated that among patients with POEMS syndrome, the bone marrow CD138+ cells (PCs) had higher levels of VEGF messenger RNA expression than did CD138− cells in the bone marrow.46
Histopathology
The diagnosis of all 4 subtypes of CD first requires an excised lymph node biopsy specimen that is consistent with classic CD histopathological findings. For ∼50 years, the nomenclature for CD histopathologic classification was split into the hyaline vascular (HV) and PC types with an intermediate “mixed” type (Figure 1).2,3 However, the features present in the HV, PC, and mixed histological subtypes occur across a spectrum rather than fitting into 3 easily definable groups. In 2017, a publication, based on the consensus work of multiple pathologists and clinicians to establish consensus diagnostic criteria for iMCD, was made (Figures 1 and 2).11 The expert panel defined the spectrum of iMCD histopathological features as follows: patients with regressed germinal centers and prominent vascularization were considered to fall on the hypervascular end of the spectrum, hyperplastic germinal centers with prominent plasmacytosis were considered to fall on the plasmacytic end of the spectrum, and patients with overlapping features of both were considered to represent mixed histopathology. These pathological findings were incorporated into consensus diagnostic criteria for iMCD (Table 1).
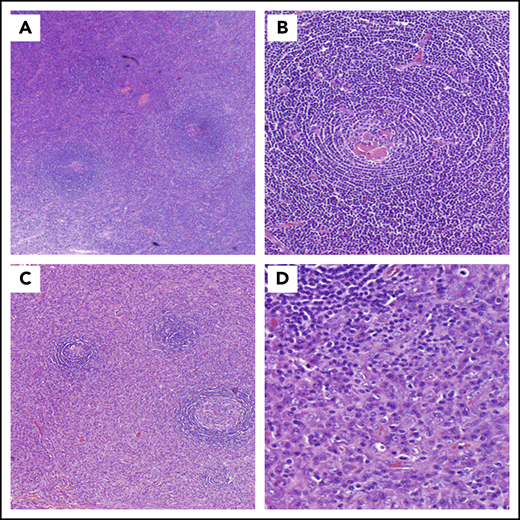
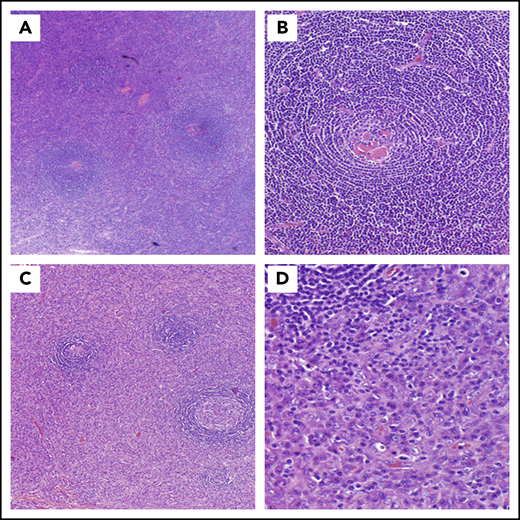
CD histopathology. (A-B) HV histopathology. (A) Low power (hematoxylin and eosin stain; original magnification ×20) and (B) high power (hematoxylin and eosin stain; original magnification ×100). Most commonly seen in UCD, these lymph nodes are often characterized by capsular fibrosis with broad fibrous bands traversing through the lymph node; an increased number of lymphoid follicles are scattered throughout the cortex and medulla with often >1 germinal center sharing the same mantle zone. Mantle zones are broad and composed of concentric rings of small lymphoid cells (“onion skin pattern”). Germinal centers are often depleted of B cells and are predominantly composed of follicular dendritic cells with prominent hyaline deposits. Sclerotic blood vessels penetrating within the germinal centers forming so-called “lollipop lesions” are observed. Follicular dendritic cells can show dysplastic features. The interfollicular region is composed of prominent high endothelial venules with plump endothelial cells, often surrounded by clusters of plasmacytoid dendritic cells and stromal proliferation. PCs, immunoblasts, and eosinophils are also part of the interfollicular infiltrate, but sheets of PCs are not seen. (C-D) PC histopathology. (C) Low power (hematoxylin and eosin stain; original magnification ×20) and (D) high power (hematoxylin and eosin stain; original magnification ×100). Lymph nodes demonstrating PC histopathology are distinguished by the presence of sheets of PCs in the interfollicular zone. The interfollicular region can also contain prominent high endothelial venules. Some eosinophils and mast cells may also be present. There is follicular/germinal center hyperplasia with sharply defined mantle zones and polarized germinal centers, with frequent mitosis and histiocytes with nuclear debris.
CD histopathology. (A-B) HV histopathology. (A) Low power (hematoxylin and eosin stain; original magnification ×20) and (B) high power (hematoxylin and eosin stain; original magnification ×100). Most commonly seen in UCD, these lymph nodes are often characterized by capsular fibrosis with broad fibrous bands traversing through the lymph node; an increased number of lymphoid follicles are scattered throughout the cortex and medulla with often >1 germinal center sharing the same mantle zone. Mantle zones are broad and composed of concentric rings of small lymphoid cells (“onion skin pattern”). Germinal centers are often depleted of B cells and are predominantly composed of follicular dendritic cells with prominent hyaline deposits. Sclerotic blood vessels penetrating within the germinal centers forming so-called “lollipop lesions” are observed. Follicular dendritic cells can show dysplastic features. The interfollicular region is composed of prominent high endothelial venules with plump endothelial cells, often surrounded by clusters of plasmacytoid dendritic cells and stromal proliferation. PCs, immunoblasts, and eosinophils are also part of the interfollicular infiltrate, but sheets of PCs are not seen. (C-D) PC histopathology. (C) Low power (hematoxylin and eosin stain; original magnification ×20) and (D) high power (hematoxylin and eosin stain; original magnification ×100). Lymph nodes demonstrating PC histopathology are distinguished by the presence of sheets of PCs in the interfollicular zone. The interfollicular region can also contain prominent high endothelial venules. Some eosinophils and mast cells may also be present. There is follicular/germinal center hyperplasia with sharply defined mantle zones and polarized germinal centers, with frequent mitosis and histiocytes with nuclear debris.
Part of the justification for altering the nomenclature within the iMCD group from HV to hypervascular was the recognition that pathologists often associate the term “hyaline vascular” with UCD even though there are iMCD patients with those histopathological features. Each variant is associated with a wide range of clinical features, so histopathology should be used to determine whether a patient has CD, but does not alone guide clinical management.
HV (or hypervascular) histopathology
Most commonly seen in UCD, lymph nodes with HV histopathology are often characterized by capsular fibrosis with broad fibrous bands traversing through the lymph node, an increased number of lymphoid follicles with regressed germinal centers, and often >1 germinal center within the same mantle zone (Figure 2A-B).3,47-49 Hypervascular is the term used to describe these features occurring in iMCD; however, iMCD patients rarely have disrupted architecture and obliterated sinuses. This hypervascular pathology is often seen among patients with iMCD-TAFRO.
PC (or plasmacytic) histopathology
Lymph nodes demonstrating PC histopathology are distinguished by the presence of sheets of PCs in the interfollicular zone and hyperplastic germinal centers (Figure 2C-D).3,50,51 PC histopathology most commonly occurs in HHV8-MCD, iMCD, and POEMS-MCD, but rarely in UCD.
In HHV8-MCD cases, there are some subtle changes not observed in other MCD types. Historically, plasmablastic histopathology has been used to describe these cases.52 Some of the reactive follicles show poorly defined mantle zones containing large immunoblasts or plasmablasts. These cells are usually positive for HHV8 latent nuclear antigen and often vIL6. The virally infected cells express monotypic (but not monoclonal) IgM-λ.29,52 These plasmablasts can form small clusters (microlymphoma) or confluent sheets (frank lymphoma); the microlymphomas can either be polyclonal or monoclonal by molecular genetic studies.29
Mixed histopathology
Lymph nodes with both HV and PC features are considered to have mixed histopathology, which can be observed in UCD and iMCD. Most commonly, these lymph nodes demonstrate extensive regressed germinal centers as well as sheet-like plasmacytosis.
Clinical findings and diagnosis
Once these classical histopathological features are observed, additional studies are needed before a formal diagnosis can be made as these histopathological features can also be seen in other disorders. If the initial lymph node biopsy is inconsistent with CD and clinical suspicion is high, an additional lymph node biopsy may be warranted. If a positron emission tomography–computed tomography (PET-CT) scan is performed, biopsy of the site with the highest standardized uptake value (SUV) is recommended, not only for obtaining a diagnostic sample but also for excluding lymphoma. Regardless of whether the CD is UCD, HHV8-MCD, POEMS-MCD, or iMCD, the median maximum SUV (SUVmax) is typically ∼3 to 8 whereas higher values would suggest lymphoma.17
Patients should have a thorough review of systems, physical examination, complete blood count, erythrocyte sedimentation rate (ESR), C-reactive protein (CRP), direct antiglobulin test (DAT), liver function tests, creatinine, serum protein electrophoresis with immunofixation, serology for HIV, urinalysis, and CT scanning of the chest, abdomen, and pelvis (or PET/CT). If there are pulmonary symptoms, pulmonary function tests should be considered. A single enlarged lymph node or region of lymph nodes on imaging suggests that a patient has UCD, whereas 2 or more regions of enlarged lymph nodes suggest that a patient has a form of MCD.
All patients with CD should be encouraged to self-enroll in the CDCN’s ACCELERATE natural history registry (#NCT02817997, www.CDCN.org/ACCELERATE) and be informed of opportunities to contribute blood samples to research (www.CDCN.org/samples).
UCD
Imaging of the chest, abdomen, and pelvis that reveals a single lymph node or region of lymph nodes along with CD histopathology is adequate to diagnose UCD in an asymptomatic patient. If B symptoms, rash, dyspnea, or peripheral neuropathy are present, further evaluation is required to derive a baseline and to exclude paraneoplastic pemphigus (PNP), bronchiolitis obliterans, and POEMS syndrome.
UCD patients typically present with either compressive symptoms or the nodes are found incidentally (Table 2).17,53 Laboratory tests are usually normal, but anemia, hypergammaglobulinemia, and elevated sedimentation rate may be present. Seventy percent to 90% of patients with UCD have HV histopathology. UCD occurs most commonly in the mediastinum, cervical regions, and abdominal/pelvic cavity, but can be found in any lymph node station. Severe complications can occur like PNP, polyneuropathy, pulmonary complications, and autoimmune hemolytic anemia.53
MCD
MCD patients exhibit lymphadenopathy in >1 lymph node station as well as a wide spectrum of clinical and laboratory abnormalities that can relapse and remit (Table 2). The most common features at presentation are constitutional symptoms, fluid accumulation, cytopenias, and liver and kidney dysfunction.6,13,54-58 The enlarged lymph nodes in MCD can occur in any lymph node station and are often vascular, appearing as well as fluorodeoxyglucose PET avid.43
Other features of MCD can include autoimmune, hemophagocytic, inflammatory, or idiopathic causes of cytopenias; hepatosplenomegaly; a multitude of renal disorders including secondary amyloidosis and membranoproliferative glomerulonephritis; peripheral neuropathy; pulmonary abnormalities such as infiltrates, restrictive lung disease, lymphoid interstitial pneumonitis, and bronchiolitis obliterans; and skin abnormalities including rash, hyperpigmentation, cherry hemangiomatosis, PNP, and Kaposi sarcoma.43 Lymphoid interstitial pneumonitis and bronchiolitis obliterans may be more common in the Asian population.
At diagnosis, assessing for HIV status and HHV8 status of the lymph node is imperative to distinguish between iMCD and HHV8-MCD. HHV8 serology is neither sensitive nor specific.15 Likewise, evaluation for possible co-occurrence with POEMS syndrome, including serum and urine protein electrophoresis with immunofixation, and neurological evaluation, is critical.
HHV8-MCD
Positive testing for HHV8 by LANA-1 of lymph node tissue and/or polymerase chain reaction for HHV8 in circulation establishes the diagnosis of HHV8-MCD in a patient with multicentric lymphadenopathy and CD histopathology.59 Baseline quantitation of plasma HHV8 is also of value for tracking disease. The hemophagocytic syndrome was present in nearly one-half of HHV8-MCD cases in 1 series.21 Three-quarters of these patients required intensive care because of hemodynamic and/or neurological failure.
Some clinical differences have been reported between HIV+ and HIV− HHV8-MCD patients. HIV+ HHV8-MCD patients were younger (42 vs 65 years of age); more likely to be white and male and have fever, splenomegaly, and hemophagocytic syndrome; and less likely to have monoclonal gammopathy or a positive DAT.17
iMCD
iMCD is clinicopathologically similar to HHV8-MCD but HHV8 is not found. Further, arthritis, cutaneous manifestations, renal disease, and lupus-like symptoms are more commonly observed in iMCD than HHV8-MCD. iMCD patients demonstrate a wide range of clinical symptoms and laboratory abnormalities as shown in Table 1. The diagnostic criteria include a lymph node consistent with iMCD histopathology, enlarged lymph nodes in at least 2 stations, and at least 2 minor criteria, with at least one of them being a laboratory criterion. Other diseases need to be ruled out by clinical history, pathologic evaluation, and additional testing as appropriate.
Further subclassification of iMCD is recommended into iMCD-TAFRO and iMCD-NOS as the presentations and acuity are quite different. For the sickest patients, the iMCD-TAFRO clinical subtype should be considered.
iMCD-TAFRO
iMCD-TAFRO describes an aggressive clinical subtype of iMCD involving thrombocytopenia, ascites, reticulin fibrosis, renal dysfunction, and organomegaly.60 It was initially described in Japan, and has also been reported among populations around the world. iMCD-TAFRO patients have hypervascularized lymph nodes, similar to the HV histopathological features described in UCD, and exhibit a different cytokine spectrum than iMCD-NOS.61,62 Whereas iMCD-NOS patients have high platelet counts and hypergammaglobulinemia, iMCD-TAFRO cases have thrombocytopenia and normal or only mildly elevated gammaglobulins.17
iMCD-NOS
iMCD-NOS describes iMCD patients who do not meet the criteria for iMCD-TAFRO. Compared with iMCD-TAFRO, iMCD-NOS cases tend to have a less aggressive clinical course, more responsiveness to corticosteroids, thrombocytosis, less frequent anasarca, lower alkaline phosphatase, and increased γ-globulin levels.10,63-65
POEMS-MCD
Occasionally, patients with HHV-8− MCD are simultaneously diagnosed with POEMS syndrome; we define this co-occurrence as POEMS-MCD and suspect that the pathologic PCs causing the POEMS syndrome are also causing the concurrent MCD. Classic POEMS syndrome is a rare paraneoplastic syndrome most often associated with osteosclerotic myeloma. Several of the defining features are in the acronym, that is peripheral neuropathy, organomegaly (hepatosplenomegaly), endocrinopathy, monoclonal gammopathy (usually λ light chain), and skin changes. Other important features not included in the acronym include sclerotic bone lesions, papilledema, extravascular volume overload, thrombocytosis, lymphadenopathy, and abnormal pulmonary function tests.66 Diagnosis of POEMS, which is required to diagnose POEMS-MCD, requires polyradiculoneuropathy, monoclonal gammopathy, and at least one of the following: sclerotic bone lesions, high VEGF, or lymph nodes consistent with CD. In many cases, the lymph nodes of classic POEMS syndrome patients have CD-like67 or classical CD histopathology, typically HV histopathological features.68,69 Interestingly, patients with POEMS-MCD without an osteosclerotic bone lesion fare much worse than those with a bone lesion.58
Peripheral neuropathy is reported to occur in ∼10% of CD patients, more commonly in MCD than UCD43,57,70 ; however, not all HHV8− MCD patients with peripheral neuropathy meet the diagnostic criteria for POEMS syndrome, and thus have POEMS-MCD. The extent and severity of peripheral neuropathy both symptomatically and objectively is less in CD patients with peripheral neuropathy but not concurrent POEMS, followed by POEMS-MCD and worst in classic POEMS without co-occurring MCD.71
When making a diagnosis of HHV8− MCD, if there is associated neuropathy, careful review of the bone images of the CT or CT/PET scans should be done looking for sclerotic bone lesions. Serum protein electrophoresis and urine protein electrophoresis is needed to search for an M-protein. Extensive endocrine testing (thyroid, adrenal, pituitary, and gonadal axes) should also be performed. A bone marrow biopsy looking for clonal PCs and megakaryocyte hyperplasia and atypia are also important. Pulmonary function tests and formal neurologic assessments should also be performed.
Differential diagnosis
The differential diagnosis of the various subtypes of CD is broad, even following a lymph node biopsy found to be consistent with CD. However, UCD, HHV8-MCD, and POEMS-MCD have a narrower differential and fewer diseases to exclude than iMCD. Few diseases other than lymphomas present with a solitary enlarged lymph node with CD-like histopathology like UCD. The presence of positive diagnostic biomarkers, HHV8 in HHV8-MCD and an M-protein in POEMS-MCD, assist with diagnosis. From a clinical perspective, multiple autoimmune diseases, such as systemic lupus erythematosus (SLE), rheumatoid arthritis, and autoimmune lymphoproliferative syndrome (ALPS), as well as acute infections and malignancies are in the differential diagnosis of iMCD. Because iMCD is so heterogeneous, the differential diagnosis differs between iMCD-TAFRO and iMCD-NOS. iMCD-TAFRO is more difficult to distinguish from SLE, myelofibrosis, acute HIV, and hemophagocytic lymphohistiocytosis, whereas iMCD-NOS is more difficult to distinguish from autoimmune lymphoproliferative syndrome, IgG4-related diseases, and Rosai-Dorfman.
Pathologically, HV histopathological features can demonstrate overlap with thymomas,1 lymphoproliferations with regressive germinal centers such as angioimmunoblastic T-cell lymphoma, and advanced phases of HIV-related lymphadenopathy.51,54,70
PC histopathological features may be seen in many other conditions, such as infections, autoimmune diseases, primary or acquired immunodeficiencies, and malignancies.51,54,70
Secondary malignancy
Secondary malignancies are not uncommon in CD. UCD patients appear to have a higher risk of developing follicular dendritic cell sarcomas43 and both Hodgkin and non-Hodgkin lymphoma.17,58 HIV-infected patients with HHV8-MCD are estimated to have a 15-fold increased frequency of lymphoma compared with an HIV-infected population without MCD.72 HIV− HHV8-MCD patients also develop malignancies, most notably lymphoma (∼15%)54,57,73-75 and Kaposi sarcoma, in as many as 50% of cases.17,54 A French collaboration reported that among HIV+ HHV8-MCD, the incidence of lymphoma in the prerituximab era was 69.6 in 1000 patient-years but fell to 4.2 in 1000 patient-years following the introduction of rituximab-based therapy.76 There is a threefold increase in malignancies in iMCD patients compared with age-matched controls.41
Therapy
UCD
The treatment decision for UCD, regardless of pathology is straightforward: surgical removal whenever possible. Complete surgical excision is almost uniformly curative with all symptoms and laboratory abnormalities returning to normal. If present, associated PNP often,77,78 but not always,79 improves within the year. Nonamyloidosis-related renal disease has also been reported to resolve within 12 months of surgical removal.80 Symptoms from associated AA amyloidosis typically improve over the ensuing years after removal of unicentric disease.81,82 Reports of lack of recovery of bronchiolitis obliterans after complete excision of UCD are difficult to interpret because it is unclear whether earlier intervention (ie, before fibrosis) would have reversed pulmonary changes.78,83
If surgery is not possible, then irradiation, embolization, or neoadjuvant therapy with rituximab or siltuximab/tocilizumab (if evidence of acute inflammatory state) should be considered.43,53,84,85 Radiation can also be effective as primary therapy,43 though it is typically reserved for select inoperable patients due to risks.
In a systematic review of 278 published UCD cases, 249 had surgical resection alone, 16 had immunosuppressive therapy alone, and 13 had a combination of surgery and immunosuppressive therapy.85 Ten-year disease-free survival was ∼90% and better among those patients who presented with peripheral lymphadenopathy. In a recent series of 71 UCD patients, only 54% had a resectable UCD lymph node at presentation.53 Overall complete response rate with surgery was 91%. Of the remaining 33 unresectable UCD patients, 19 had neoadjuvant therapy (eg, steroids, alkylators, rituximab, tocilizumab, embolization) with 7 going on to have a resection. There were 4 complete responses and 14 partial responses. Eight patients were treated with radiotherapy. Four patients had complete responses and 4 had partial responses. In toto, 11 unresectable UCD patients remained stable long-term on active surveillance without any therapy. This is an interesting observation, but one must be vigilant about secondary malignancies and progression of associated paraneoplastic entities like PNP and bronchiolitis obliterans.
MCD
Most treatment data are from case reports and series, many of which did not historically specify the subtype of MCD, making them a mélange of HHV8-MCD, POEMS-MCD, iMCD-NOS, and iMCD-TAFRO cases. Fortunately, there are several more recent series in which several of these distinctions are made.10,17,41,63,86
There are also limited data available related to outcomes and prognosis. In an excellent review of 253 patients with CD, Oksenhendler et al report on the overall survival (OS) of CD patients at his institution in France over a 20-year period.17 The estimated 5-year OS was as follows: iMCD, 100%; HIV− HHV8-MCD, 89%; and HIV+ HHV8-MCD, 65%. The estimated OS for iMCD was extraordinarily good, but only 2 of the 27 patients had iMCD-TAFRO. In an older case series, before the approval of anti–IL-6 therapy, the 5-year OS for HIV− (presumed HHV8−) MCD was 65%.58
HHV8-MCD
Rituximab-based therapy has dramatically improved 5-year OS for HHV8-MCD from 33% to 90%.87 Even though HHV8-infected plasmablasts frequently do not express high levels of CD20, rituximab has been successfully applied in multiple case series and 3 open-label studies.86 Rituximab is used as a single agent weekly for 4 weeks in patients with adequate performance status and absence of hemophagocytic syndrome, hemolytic anemia, or end-organ damage. High-risk patients were treated with rituximab and etoposide weekly for 4 weeks.88,89 With such an approach, 95% of patients achieved clinical remission, 5-year OS was 92%, and 5-year relapse-free survival was 82%. All patients were successfully retreated at relapse with rituximab-based therapy. Rituximab-based approaches have also reduced the risk of HHV8-associated lymphomas.76,86 With the use of rituximab, however, worsening Kaposi sarcoma occurs in one- to two-thirds of patients with baseline Kaposi sarcoma.88,89 The combination of rituximab and liposomal doxorubicin every 3 weeks appears to attenuate Kaposi sarcoma exacerbation.90 In 1 pilot study, high-dose valganciclovir and zidovudine combination yielded clinical response rates approaching 90%, but progression-free survival was only 6 months.91 Rituximab with or without etoposide appears to be effective in HHV8-MCD, regardless of HIV status.17
POEMS-MCD
For those patients with concurrent POEMS syndrome and MCD who have osteosclerotic lesions and predominant peripheral neuropathy symptoms treated with standard myeloma therapy, preferably high-dose chemotherapy with autologous stem cell transplant (ASCT) is warranted. For patients who are not candidates for ASCT, other myeloma-type therapies like melphalan, cyclophosphamide, lenalidomide, thalidomide, bortezomib, carfilzomib, and daratumumab can be considered, understanding that most of these recommendations come from small case series.66,92
For patients with POEMS-MCD without bone lesions, there are even less data. If there is high IL-6, siltuximab and rituximab can be considered as outlined for iMCD, but moving to PC-directed immune-modulatory therapy should also be considered.
iMCD
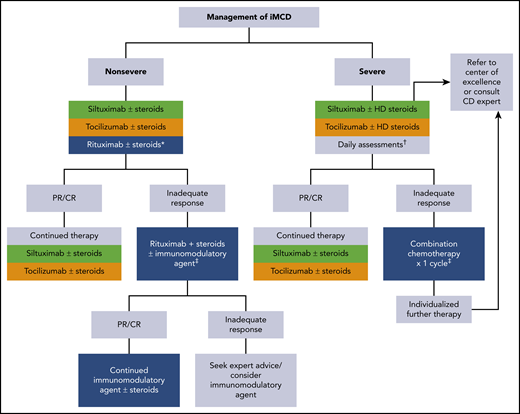
Consensus guidance from van Rhee et al outlines treatment recommendations for iMCD (Figure 3).84 When choosing therapy for patients with iMCD, the severity of disease (Table 4) must be considered.
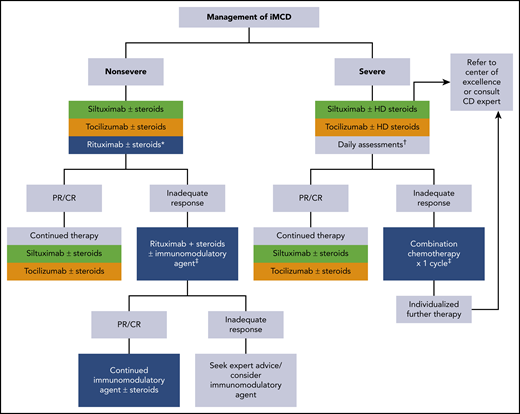
Consensus guidance for the treatment of idiopathic multicentric CD. Adapted from van Rhee et al.84 iMCD patients should be stratified for disease severity per Table 4. *For patients with mild symptomatology, a limited course of rituximab is an alternative option. †Accelerated weekly dosing of anti-IL-6 therapy is recommended for the first month along with daily assessment of the patient's status. If organ dysfunction worsens at any time, initiation of combination chemotherapy should be considered (see text). ‡Examples of therapies are listed in Table 5. Green is category 1 evidence: based on high-level evidence; there is uniform consensus that the intervention is appropriate. Gold is category 2A evidence: based on lower-level evidence; there is uniform consensus that the intervention is appropriate. Blue is category 2B evidence: based on lower-level evidence; there is consensus that the intervention is appropriate. CR, complete response; HD steroids, high-dose steroids; mAb, monoclonal antibody; PR, partial response.
Consensus guidance for the treatment of idiopathic multicentric CD. Adapted from van Rhee et al.84 iMCD patients should be stratified for disease severity per Table 4. *For patients with mild symptomatology, a limited course of rituximab is an alternative option. †Accelerated weekly dosing of anti-IL-6 therapy is recommended for the first month along with daily assessment of the patient's status. If organ dysfunction worsens at any time, initiation of combination chemotherapy should be considered (see text). ‡Examples of therapies are listed in Table 5. Green is category 1 evidence: based on high-level evidence; there is uniform consensus that the intervention is appropriate. Gold is category 2A evidence: based on lower-level evidence; there is uniform consensus that the intervention is appropriate. Blue is category 2B evidence: based on lower-level evidence; there is consensus that the intervention is appropriate. CR, complete response; HD steroids, high-dose steroids; mAb, monoclonal antibody; PR, partial response.
For all patients, regardless of disease severity, the algorithm starts with anti–IL-6–directed therapy. The only drug tested in a randomized trial for iMCD is siltuximab.93 Siltuximab, an anti–IL-6 antibody, is the only US Food and Drug Administration (FDA)-approved treatment of iMCD. In the registration study of 79 patients, 34% of patients in the siltuximab arm had durable symptomatic and tumor responses; the placebo arm had none. Although there was a trend toward a higher response rate among patients with high IL-6 levels, some iMCD patients with low or normal values responded to siltuximab whereas some patients with high levels did not.94 Patients in the trial with high immunoglobulins, CRP, and fibrinogen and low hemoglobin were most likely to respond to siltuximab.95 Data from the use of tocilizumab, an anti–IL-6 receptor antibody approved for iMCD in Japan based on a single-arm open-label study, would suggest that relapses occur upon cessation of therapy.96 Overall, both therapies are well tolerated with the most common side effects being hyperlipidemia, mild thrombocytopenia, and pruritus. IL-6 levels become uninterpretable while patients are on active anti–IL-6 therapy and cannot be used to monitor response for 18 to 24 months following the last dose of siltuximab. Moreover, lymph node responses are often delayed in patients receiving anti–IL-6 antibody monotherapy compared with chemotherapy. The most reliable measures are hemoglobin, ESR, CRP, albumin, and clinical symptomology.84
For patients with nonsevere iMCD who do not respond to IL-6 blockade, many therapies can be tried, and it is difficult to recommend one over another. The general rule, however, is to avoid cytotoxic chemotherapy as long as the patient does not have severe disease with progressive organ dysfunction. Options that can be considered are shown in Table 5 and include corticosteroids, rituximab, thalidomide, lenalidomide, bortezomib, cyclosporine, sirolimus, interferon, as well as others.38,41,58,63,84,97,98 Nearly one-half of patients will demonstrate some temporary improvement with corticosteroids, but relapses occur, and long-term high-dose corticosteroids are associated with significant morbidity. Rituximab can induce responses in some patients41 and is considered as an alternative first-line option for those patients with mild iMCD symptomatology and as second line for anti–IL-6 failures. In a retrospective analysis, the progression-free survival was superior among those patients treated with siltuximab as compared with either rituximab/rituximab-based therapies or cytotoxic chemotherapy regimens63 ; a caveat is that these analyses were not corrected for disease severity. Thalidomide has also been used with some success in iMCD patients. A phase 2 study of oral thalidomide, cyclophosphamide, and prednisone in 25 newly diagnosed iMCD patients demonstrated that 48% of patients achieved the primary end point of durable tumor and symptomatic response for at least 24 weeks.98 The estimated 1-year progression-free and overall survival was 60% and 88%, respectively. High-dose chemotherapy with ASCT has been rarely reported with variable success that appears inferior to what is seen in patients with POEMS syndrome.99
For patients with severe disease, which may or may not meet the criteria for iMCD-TAFRO, anti–IL-6–directed therapy is still indicated first line but dosing should occur weekly for the first month and concurrent high-dose corticosteroids (methylprednisolone 500 mg daily) are initially indicated. Daily assessment should occur, with an eye toward starting cytotoxic chemotherapy if progressive organ dysfunction occurs at any time.84 Cytotoxic regimens that one would consider for lymphoma, myeloma, or hemophagocytic lymphohistiocytosis have been used even among patients in the intensive care unit. These intensive regimens may be necessary to break the cytokine/chemokine storm in these patients. Overall responses for these aggressive regimens are in the order of 75%, but relapses are common. Maintenance strategies are designed on an ad hoc basis.
Because iMCD-TAFRO is a relatively newly recognized subtype, there is even less information about the best therapies for these patients. They can respond to anti-IL-6 therapies, calcineurin inhibitors like tacrolimus and other immune-modulatory drugs like sirolimus, but data are mostly anecdotal.100-102 A clinical trial is currently evaluating sirolimus in iMCD patients who are relapsed or refractory to anti–IL-6 therapy at the University of Pennsylvania and the University of Arkansas for Medical Sciences (NCT03933904).
Acknowledgment
The authors acknowledge the major contributions of the Castleman Disease Collaborative Network, especially its scientific advisory board.
Authorship
Contribution: A.D. wrote the first draft of manuscript; and D.C.F. revised and added content to the manuscript.
Conflict-of-interest disclosure: A.D. received research dollars from Intellia, Pfizer, Celgene, Takeda, and Alnylam. D.C.F. receives funding from Janssen Pharmaceuticals and serves on the Board of Directors for the Castleman Disease Collaborative Network
Correspondence: Angela Dispenzieri, Mayo Clinic, 200 First St SW, Rochester, MN 55905; e-mail: dispenzieri.angela@mayo.edu.