Key Points
MRD assessment in t(8;21) AML allows identification of patients at high relapse risk at defined time points during treatment and follow-up.
MRD− after treatment is the most favorable factor for relapse risk and survival, and serial MRD analyses define cutoffs predicting relapse.

Abstract
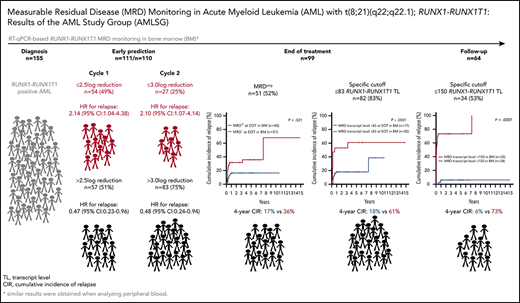
We performed serial measurable residual disease (MRD) monitoring in bone marrow (BM) and peripheral blood (PB) samples of 155 intensively treated patients with RUNX1-RUNX1T1+ AML, using a qRT-PC–based assay with a sensitivity of up to 10−6. We assessed both reduction of RUNX1-RUNX1T1 transcript levels (TLs) and achievement of MRD negativity (MRD−) for impact on prognosis. Achievement of MR2.5 (>2.5 log reduction) after treatment cycle 1 and achievement of MR3.0 after treatment cycle 2 were significantly associated with a reduced risk of relapse (P = .034 and P = .028, respectively). After completion of therapy, achievement of MRD− in both BM and PB was an independent, favorable prognostic factor in cumulative incidence of relapse (4-year cumulative incidence relapse: BM, 17% vs 36%, P = .021; PB, 23% vs 55%, P = .001) and overall survival (4-year overall survival rate BM, 93% vs 70%, P = .007; PB, 87% vs 47%, P < .0001). Finally, during follow-up, serial qRT-PCR analyses allowed prediction of relapse in 77% of patients exceeding a cutoff value of 150 RUNX1-RUNX1T1 TLs in BM, and in 84% of patients exceeding a value of 50 RUNX1-RUNX1T1 TLs in PB. The KIT mutation was a significant factor predicting a lower CR rate and inferior outcome, but its prognostic impact was outweighed by RUNX1-RUNX1T1 TLs during treatment. Virtually all relapses occurred within 1 year after the end of treatment, with a very short latency from molecular to morphologic relapse, necessitating MRD assessment at short intervals during this time period. Based on our data, we propose a refined practical guideline for MRD assessment in RUNX1-RUNX1T1+ AML.
Introduction
Acute myeloid leukemia (AML) with t(8;21)(q22;q22.1); RUNX1-RUNX1T1 is a distinct entity within the category “AML with recurrent genetic abnormalities” of the World Health Organization classification.1 Translocation t(8;21) leading to the formation of the RUNX1-RUNX1T1 gene fusion is considered to be a favorable AML subset in the 2017 risk stratification by the European LeukemiaNet (ELN).2 Although most patients achieve complete remission (CR) after intensive chemotherapy, relapse occurs in ∼50% of patients and is associated with a poor prognosis.3-6
Quantification of RUNX1-RUNX1T1 transcript levels (TLs) by quantitative reverse transcription-polymerase chain reaction (qRT-PCR) for monitoring of measurable residual disease (MRD) provides a sensitive assay for the identification of patients at higher risk of relapse. However, standardizing MRD assays and choosing the right time points to inform treatment remained challenging. Previously, we had identified clinically relevant checkpoints for increased relapse risk in AML with inv(16)(p13.1q22) or t(16;16)(p13.1;q22): CBFB-MYH11.7 In the past few years, several studies of MRD monitoring in RUNX1-RUNX1T1+ AML described distinct time points of prognostic relevance. In a 2016 study from the French AML Intergroup of 94 patients with RUNX1-RUNX1T1+ AML, achievement of complete molecular remission (CMR) ([RUNX1-RUNX1T1/ABL1] × 100; <0.001%) in peripheral blood (PB) after the end of consolidation therapy was predictive of a lower likelihood of subsequent relapse.8 Another study of 96 RUNX1-RUNX1T1+ AML patients showed that a 3-log TL reduction after consolidation cycle 1 correlated with decreased relapse risk.9 Furthermore, the United Kingdom Medical Research Council group reported that different prognostic MRD thresholds for bone marrow (BM) and PB obtained at defined time points are significantly associated with outcome.10
Despite the heterogeneity of published studies and diverse definitions of MRD negativity (MRD−) and the cutoffs that have been used, there is broad consensus that MRD persistence is associated with increased risk of relapse, resulting in an inferior outcome. The 2017 ELN recommendations acknowledged the increasing importance of MRD monitoring by introducing a new response criterion (ie, CR without measurable residual disease [CRMRD−]). In accordance, the 2018 consensus document from the ELN MRD Working Party aims to address some of the key methodological and clinical issues and provides recommendations for MRD in clinical practice.11
The objective of our study was to assess the prognostic impact of qRT-PCR–based MRD monitoring in BM and PB in a large cohort of 155 homogeneously treated and clinically well-annotated patients with t(8;21)-AML at defined time points.12
Patients and methods
Patients and treatment
One-hundred fifty-five patients (median age, 48 years; range, 18-76; ≤60 years, n = 126; >60 years, = 29) with RUNX1-RUNX1T1+ AML were included based on the availability of a sample at diagnosis and at least at 2 subsequent time points during the disease. Overall 2297 samples were analyzed (BM, n = 1182; PB, n = 1115; supplemental Table 1; available on the Blood Web site). One-hundred thirty-nine patients were enrolled in 1 of 6 treatment trials of the German-Austrian AML Study Group (AMLSG): AML HD93 (n = 1),13 AML HD98A (n = 14),14 AMLSG 06-04 (n = 4),15 AMLSG 07-04 (n = 43),16 AMLSG 11-08 (n = 32),17 and AMLSG 21-13 (NCT02013648; n = 45); 16 patients received intensive treatment according to the standard of care.18 Patient characteristics are presented in Table 1; cohorts were comparable regarding baseline characteristics and outcomes (supplemental Table 2). Fifty-three patients received dasatinib in addition to intensive chemotherapy. The study was conducted in accordance with the Declaration of Helsinki. Written informed consent for treatment and genetic testing was obtained from all patients.
Quantification of RUNX1-RUNX1T1 TLs by qRT-PCR
Mononuclear cells were isolated by Ficoll density gradient from BM and PB specimens. For quantification of RUNX1-RUNX1T1 TLs, qRT-PCR was performed with a QuantStudio 12K Flex Real-Time PCR System (Applied Biosystems, Foster City, CA). Primers and probes were selected according to the standard protocols of A Europe Against Cancer Program19 ; β2-microglobulin (B2M) was used as housekeeping gene, as described.7,20 Further information on sample preparation and qRT-PCR is provided in the supplemental Appendix.
Molecular analyses
Mutation testing for NRAS (exons 1 and 2), KIT (exons 8 and 17), ASXL2 (exons 11 and 12), and FLT3 internal tandem duplications and tyrosine kinase domain mutations was performed as previously reported.21-23
Statistical analyses
Details of the statistical analyses are provided in the supplemental Appendix.
Results
Patient characteristics
All 155 patients received anthracycline- and standard-dose, cytarabine-based induction therapy. After the first cycle of chemotherapy, 119 (77%) patients achieved CR and 3 (2%) patients had refractory disease (RD); there was no early death. After the second cycle of chemotherapy (induction 2, n = 87; consolidation 1, n = 68), 152 (98%) patients achieved CR. One-hundred forty-five patients received consolidation therapy with repetitive cycles of high-dose cytarabine (n = 145), and allogeneic and autologous hematopoietic cell transplantations (HCTs) were performed in the first CR in 8 and 2 patients, respectively. The median follow-up time for survival was 4.0 years. Of the 155 patients, 37 (23.9%) died. Median overall survival (OS), event-free survival (EFS), and relapse-free survival (RFS) were 11.2 years (95% CI, 2.5-19.8), 11.2 years (95% CI, 4.0-18.3), and 11.0 years (95% CI, 3.9-18.0), respectively. The 4-year survival rates for OS, EFS, and RFS were 75%, 64%, and 64%, respectively. Forty-five patients (29%) relapsed, and 42 (93%) of these relapses occurred within 18 months after diagnosis (supplemental Figure 1). Of the 45 relapsed patients, 32 patients underwent allogeneic HCT (14 in second CR and 18 with active disease).
RUNX1-RUNX1T1 TLs at diagnosis
Overall, 280 samples (BM, n = 145; PB, n = 135) from 155 patients were available at diagnosis (with paired BM/PB samples in n = 125 patients). The median TLs were 228 067 in BM (range 3109-14 440 420) and 223 940 in PB (range 2910-10 380 408). Pretreatment RUNX1-RUNX1T1 TL correlated with PB blast counts (BM, ρ [Spearman] = 0.264, P = .003; PB, ρ = 0.310, P = .001); in addition, the PB TL correlated with white blood cell (WBC) count (ρ = 0.200; P = .024) and inversely with age (ρ = −0.195; P = .023). There was no correlation with gender, platelet counts, lactate dehydrogenase (LDH) level, BM blast counts, or gene mutations in KIT, FLT3, NRAS, or ASXL2.
Pretreatment RUNX1-RUNX1T1 TLs as a log10-transformed continuous variable did not impact OS (BM, P = .253; PB, P = .628), EFS (BM, P = .428; PB, P = .456), RFS (BM, P = .430; PB, P = .455), or cumulative incidence of relapse (CIR: BM, P = .373; PB, P = .607).
Prognostic impact of RUNX1-RUNX1T1 TLs during therapy
Response to induction chemotherapy
There was no correlation with response of RUNX1-RUNX1T1 TLs, age, WBC, LDH, BM blast count, or mutation in the NRAS, FLT3, or ASXL2 genes. KIT mutation was the only variable predictive of lower CR rate after the first induction cycle (odds ratio [OR], 0.34; 95% confidence interval [CI], 0.15-0.80; P = .014), retaining its impact in multivariate analysis (OR, 0.28; 95% CI, 0.11-0.76; P = .012; supplemental Table 3).
Survival analysis
We performed Cox regression analyses to determine the prognostic impact of RUNX1-RUNX1T1 TLs, as well as their reduction in relation to diagnosis in BM and PB obtained at the following time points: after the first cycle (cycle 1) of chemotherapy (BM, n = 111; PB, n = 91), after the second cycle (cycle 2) of chemotherapy (BM, n = 110; PB, n = 101), and at the end of treatment (EOT) (BM, n = 99; PB, n = 86). In general, at all 3 time points, higher RUNX1-RUNX1T1 TLs and less MRD reduction in both BM and PB were associated with higher risk of relapse and inferior OS, with the exception of only a very few values that did not reach statistical significance (Table 2).
After 1 cycle of chemotherapy
The median TL at this time point was 825 (range, 0-460 362) in BM and 194 (range, 0-57 662) in PB. The median log10 reduction of RUNX1-RUNX1T1 TLs in relation to baseline was −2.53 for BM and −2.89 for PB. Therefore, we determined the prognostic impact of TL reduction comparing >2.5-log reduction (MR2.5; BM, n = 57 [51%]; PB, n = 59 [65%]) with ≤2.5-log reduction (BM, n = 54 [49%]; PB, n = 32 [35%]). Achievement of MR2.5 was associated with a lower 4-year CIR (BM, 22% vs 43%, P = .034; PB, 19% vs 51%; P = .008) and on trend with a superior 4-year OS rate (BM, 77% vs 61%, P = .078; PB, 84% vs 66%; P = .083). MRD− was achieved in only 2 patients in BM and in 4 patients in PB, precluding meaningful outcome analyses.
After 2 cycles of chemotherapy
The median TL at this time point was 47 (range, 0-11 183) in BM and 3 (range, 0-14 746) in PB. The median log10 reduction in RUNX1-RUNX1T1 TL in relation to baseline was −3.79 for BM and −4.80 for PB. Analogous to the study of the French AML Intergroup,9 we examined TL reduction after 2 cycles comparing >3-log reduction (MR3.0; BM, n = 83 [75%]; PB, n = 89 [88%]) vs ≤3-log reduction (BM, n = 27 [25%]; PB, n = 13 [12%]). CIR was significantly lower if MR3.0 was achieved in BM (4-year CIR, 28% vs 51%; P = .028) or PB (4-year CIR, 29% vs 54%; P = .036); there was no impact on OS. In multivariate Cox regression, MR3.0 retained its significance for CIR in both BM (hazard ratio [HR], 0.48; 95% CI, 0.24-0.98; P = .043) and PB (HR, 0.35; 95% CI, 0.14-0.85; P = .021). Other variables had no impact on CIR (supplemental Table 4). MRD− after 2 cycles was achieved in BM in 28 of 110 (25%) patients and in PB in 48 of 101 (48%) patients. There was no correlation with any clinical end point for achievement of BM and PB MRD−.
At EOT
The median TL at this time point was 0 (range, 0-132 971) in BM and 0 (range, 0-139 495) in PB. MRD− at EOT was achieved in BM in 51 of 99 (52%) patients and in PB in 64 of 86 (74%) patients. MRD− at EOT in both BM and PB samples predicted superior 4-year rates of CIR (BM, 17% vs 36%; P = .021; PB, 23% vs 55%; P = .001) and OS (BM, 93% vs 70%; P = .007; PB, 87% vs 47%; P < .0001; Figure 1). Moreover, using maximally selected Gray’s statistic for competing risks, we defined specific MRD cutoff values at EOT that were associated with a low risk of relapse. MRD levels below 83 RUNX1-RUNX1T1 TLs in BM and 5 in PB predicted superior 4-year rates of CIR (BM, 18% vs 61%; P < .0001; PB, 23% vs 65%; P < .0001; supplemental Figure 2).
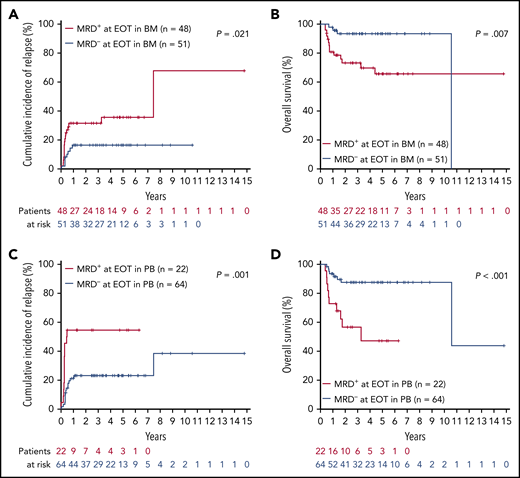
Relapse risk and outcome for patients in CR according to MRD status at EOT. CIR (A) and OS (B) in BM (negative vs any positive RUNX1-RUNX1-T1 TL value). CIR (C) and OS (D) in PB.
Relapse risk and outcome for patients in CR according to MRD status at EOT. CIR (A) and OS (B) in BM (negative vs any positive RUNX1-RUNX1-T1 TL value). CIR (C) and OS (D) in PB.
To evaluate the impact of RUNX1-RUNX1T1 TLs at EOT, 3 multivariate Cox regression models were performed: 1 including log10-transformed BM or PB TLs at EOT, 1 including BM or PB MRD cutoffs at EOT, and 1 comprising BM or PB MRD− at EOT. In all models, higher RUNX1-RUNX1T1 TLs, MRD cutoff values, and MRD− at EOT retained their prognostic significance for OS and risk of relapse (Table 3). This was also true when the analysis was stratified according to additional dasatinib treatment (supplemental Table 5). Another variable predicting inferior outcome was KIT mutation (KITmut), but this impact was not consistent.
Impact of concurrent KIT mutations
KITmut was associated with a lower CR rate (OR, 0.34; 95% CI, 0.15-0.80; P = .014); inferior OS (HR, 2.21; 95% CI, 1.12-4.37; P = .022), EFS (HR, 2.17; 95% CI, 1.24-3.82; P = .007), and RFS (HR, 2.08; 95% CI, 1.18-3.66; P = .011); and a trend toward higher CIR (HR, 1.82; 95% CI, 0.97-3.39; P = .061).
We next evaluated the impact of concurrent KITmut on RUNX1-RUNX1T1 TL kinetics in BM and PB. Median BM log10-transformed TLs were not significantly different between KIT wild-type (KITWT) and KITmut patients at diagnosis (5.38 vs 5.34; P = .720), after cycle 1 (2.84 vs 3.15; P = .072), and after cycle 2 (1.57 vs 1.96; P = .144), but the difference reached statistical significance at EOT (−0.48 vs 1.41; P = .039; Figure 2A). There were no differences for TLs in PB. However, when analyzing log10 TL reduction, we observed a higher reduction in BM TLs in KITWT vs KITmut cases after cycle 1 (−2.77 vs −2.31; P = .045) and at EOT (−5.35 vs −4.08; P = .043; Figure 2B). There were no differences in log10 reduction in PB. Furthermore, KITmut status was associated with achievement of BM MRD− at EOT. Only 8 of 23 (35%) patients with KITmut became MRD−, compared with 38 of 66 (58%) patients with KITWT (P = .05).
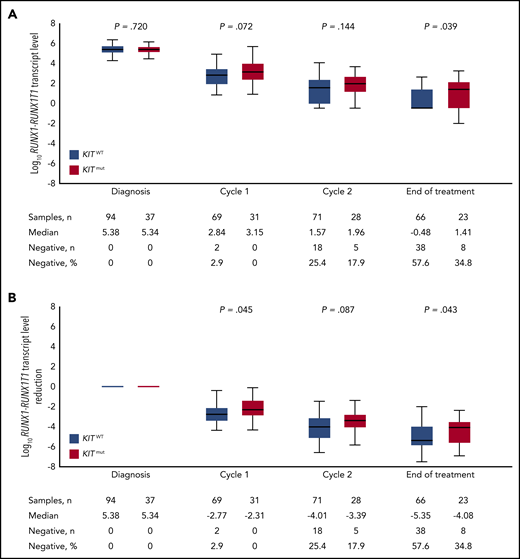
Impact of KIT mutation status on BM MRD kinetics. Log10RUNX1-RUNX1T1 TLs (A) and the reduction of RUNX1-RUNX1T1 TLs (B) are shown during the course of treatment.
Impact of KIT mutation status on BM MRD kinetics. Log10RUNX1-RUNX1T1 TLs (A) and the reduction of RUNX1-RUNX1T1 TLs (B) are shown during the course of treatment.
Impact of additional dasatinib treatment
Because 53 patients received intensive chemotherapy combined with dasatinib, we evaluated the impact of additional dasatinib treatment on RUNX1-RUNX1T1 TL kinetics in BM and PB and on outcome. Additional dasatinib treatment was associated with lower RUNX1-RUNX1T1 TLs and stronger MRD reduction at EOT in both BM (P = .023 and P = .037, respectively) and PB (P = .014 and P = .019, respectively; supplemental Figure 3), as well as with achievement of MRD− at EOT in PB (28 of 31 vs 36 of 55; P = .011; supplemental Table 6). However, additional dasatinib treatment had no impact on OS or CIR (supplemental Table 7).
MRD monitoring during follow-up
To assess the risk of relapse after completion of therapy, we performed serial measurements during the posttreatment period in 531 BM and 537 PB samples obtained from 119 and 112 patients, respectively. We followed a predefined schedule of every 3 months, per protocol, and the median time interval of sample acquisition was 3.3 months for both BM and PB. For BM, MRD− was achieved in 51 patients at EOT and in an additional 33 patients (23 of 48 MRD+ patients at EOT and 10 of 20 patients without available MRD assessment at EOT) during follow-up. Notably, 9 of the 84 (11%) patients achieving MRD− relapsed, whereas the percentage of relapse in patients not achieving MRD− was significantly higher (26 of 35 [74%] patients; P < .0001; Table 4). Almost identical data were observed for PB: MRD− was achieved in 64 patients at EOT and in an additional 29 patients (9 of 22 MRD+ patients at EOT and 20 of 26 patients without available MRD assessment at EOT) during follow-up. Notably, 19 of the 93 (20%) patients achieving MRD− relapsed compared with 14 of the 19 (74%) patients not achieving MRD− (P < .0001; Table 4). Repetitive MRD− (at least 2 consecutive assessments) during follow-up was achieved in 52 patients in BM and in 68 patients in PB. Of the 52 patients with repetitive MRD− in BM, 51 (98%) remained in CR and only 1 (2%) relapsed, whereas in patients without repetitive BM MRD− (n = 67), only 34 (51%) remained in CR and 33 (49%) relapsed (P < .0001; Table 4). Of the 68 patients achieving repetitive MRD− in PB, 65 (96%) remained in CR and only 3 (4%) relapsed. In contrast, in patients without repetitive PB MRD− (n = 44), only 14 (32%) remained in CR and 30 (68%) relapsed (P < .0001; Table 4).
We then evaluated the prognostic impact of MRD conversion from MRD− to MRD+. During follow-up, 25 patients converted to MRD+ in the BM sample within 3.77 months (median time from last MRD− to first MRD+ sample; range, 2.83-18.2 months); 6 patients (24%) experienced relapse with a steep rise in MRD kinetics (from MRD− to a median TL of 18 507.14; range, 186.35-1 310 886.32) within a median time of 3.4 months (range, 2.8-7.7). Of the 19 patients without clinical relapse (TL range, 0.01-635), 13 again became MRD− without any treatment intervention.
In PB, 25 patients converted to MRD+ within 3.3 months (median time from last MRD− to first MRD+ sample; range, 1-11.4 months), and 16 (64%) of these patients relapsed with strong, increasing MRD kinetics (from MRD− to a median TL of 1269; range, 2.97-373 553.29) within a median time of 3.2 months (range, 1-11.4 months). Of the 9 patients with no clinical relapse (TL range; 0.01-271), 5 again became MRD− without any treatment intervention. The median time from MRD conversion to morphological relapse was only 0.8 months (range, 0-5.1.7) and 1.6 months (range, 0-84.6) in BM and PB, respectively (Figure 3).
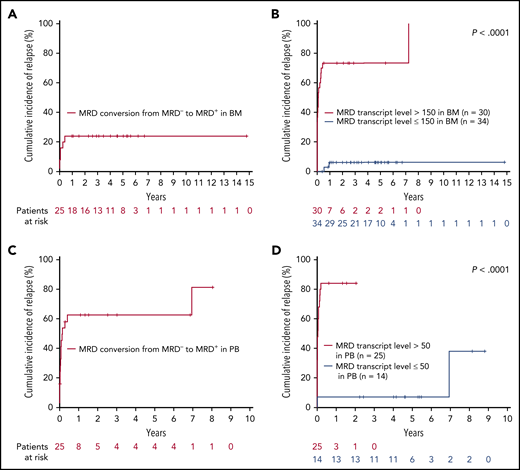
CIR during follow-up, according to MRD conversion (from MRD−to MRD+) and defined MRD cutoffs in BM and PB. (A) CIR of 25 patients with MRD conversion in BM. (B) CIR of 64 patients according to the MRD cutoff exceeding 150 RUNX1-RUNX1T1 TL/106B2M copies in at least 1 BM follow-up sample obtained in the posttreatment period. (C) CIR of 25 patients with MRD conversion in PB. (D) CIR of 39 patients according to MRD cutoff exceeding 50 RUNX1-RUNX1T1 TL/106B2M copies in at least 1 PB follow-up sample obtained in the posttreatment period. Time to relapse is calculated from the first sample, with a RUNX1-RUNX1T1 TL >150 (BM) or >50 (PB) RUNX1-RUNX1T1 TL/106B2M copies up to relapse and, in cases not exceeding these thresholds, from the first sample with increasing MRD level.
CIR during follow-up, according to MRD conversion (from MRD−to MRD+) and defined MRD cutoffs in BM and PB. (A) CIR of 25 patients with MRD conversion in BM. (B) CIR of 64 patients according to the MRD cutoff exceeding 150 RUNX1-RUNX1T1 TL/106B2M copies in at least 1 BM follow-up sample obtained in the posttreatment period. (C) CIR of 25 patients with MRD conversion in PB. (D) CIR of 39 patients according to MRD cutoff exceeding 50 RUNX1-RUNX1T1 TL/106B2M copies in at least 1 PB follow-up sample obtained in the posttreatment period. Time to relapse is calculated from the first sample, with a RUNX1-RUNX1T1 TL >150 (BM) or >50 (PB) RUNX1-RUNX1T1 TL/106B2M copies up to relapse and, in cases not exceeding these thresholds, from the first sample with increasing MRD level.
Finally, we sought to establish an arbitrary, but clinically meaningful, MRD cutoff for patients with at least 1 MRD+ sample during the follow-up period. In BM, RUNX1-RUNX1T1 TL >150 identified 23 of 30 (77%) patients with subsequent relapse, whereas 34 patients with ≤150 TL of only 2 (6%) relapsed (P < .0001; Figure 3). Thirty-nine patients were available for PB analysis: 21 of 25 (84%) patients with MRD levels >50 relapsed, whereas relapse occurred in only 2 of 14 (14%) patients with TL ≤50 (P < .0001; Figure 3). The median time to relapse was only 0.9 months (range, 0-87.7) and 0.3 months (range, 0-24.9) in cases exceeding the respective cutoffs in BM and PB, respectively. Notably, these cutoffs were valid irrespective of dasatinib treatment (supplemental Figure 4).
Paired BM and PB analysis
To determine whether PB could provide similar prognostic information as BM, we compared 680 paired samples at diagnosis (n = 125), after cycle 1 (n = 80), after cycle 2 (n = 86), at EOT (n = 78), and during follow-up (n = 311; Figure 4A). At diagnosis, median RUNX1-RUNX1T1 TLs tended to be slightly higher in BM (241 585 RUNX1-RUNX1T1/106B2M copies) than in PB (219 368 RUNX1-RUNX1T1/106B2M copies; P = .072); TLs were significantly higher in BM compared with PB after cycle 1 (P = .008), after cycle 2 (P < .001), at EOT (P = .002), and during follow-up (P < .001; Figure 4B). RUNX1-RUNX1T1 TLs in BM and PB correlated well (r = 0.87; P < .0001; Figure 4C). However, 2.5%, 26.7%, 26.9%, and 24.8% of all pairs were discrepant (BM+/PB− or BM−/PB+) after cycles 1 and 2, at EOT, and during follow-up, respectively (Figure 4D). Of 104 negative PB samples obtained during induction and consolidation therapy, 46 samples (44%) still showed RUNX1-RUNX1T1 TLs in BM. In the posttreatment period, this fraction decreased to 28% (77 BM+/276 PB− pairs; P = .003; Figure 4D). Of note, RUNX1-RUNX1T1 TLs in all but 4 of the 77 (5.2%) BM+ samples were below the cutoff of 150 RUNX1-RUNX1T1/106B2M copies.

Analysis of paired BM and PB samples in 125 patients. (A) Histogram plot of paired BM and PB RUNX1-RUNX1T1 TLs during therapy and the posttreatment period of the 125 patients, illustrating sample acquisition, interindividual RUNX1-RUNX1T1 TLs, intraindividual correlation of BM and PB TLs, and the MRD course of each patient. Color-coded histograms represent paired RUNX1-RUNX1T1 TLs in BM and PB (y-axis) at the indicated time points (x-axis) for each patient (z-axis). Missing samples are denoted by a corresponding blank area. (B) RUNX1-RUNX1T1 TL kinetics of paired BM and PB samples during therapy and follow-up. (C) Spearman rank correlation of RUNX1-RUNX1T1 TLs of all 680 paired BM and PB samples obtained during therapy and in the posttreatment period. (D) Distribution of paired samples according to their qRT-PCR status (positive/negative) in BM and PB.
Analysis of paired BM and PB samples in 125 patients. (A) Histogram plot of paired BM and PB RUNX1-RUNX1T1 TLs during therapy and the posttreatment period of the 125 patients, illustrating sample acquisition, interindividual RUNX1-RUNX1T1 TLs, intraindividual correlation of BM and PB TLs, and the MRD course of each patient. Color-coded histograms represent paired RUNX1-RUNX1T1 TLs in BM and PB (y-axis) at the indicated time points (x-axis) for each patient (z-axis). Missing samples are denoted by a corresponding blank area. (B) RUNX1-RUNX1T1 TL kinetics of paired BM and PB samples during therapy and follow-up. (C) Spearman rank correlation of RUNX1-RUNX1T1 TLs of all 680 paired BM and PB samples obtained during therapy and in the posttreatment period. (D) Distribution of paired samples according to their qRT-PCR status (positive/negative) in BM and PB.
Discussion
In this study, we report the results of our prospective longitudinal MRD monitoring in 155 intensively treated adult patients with RUNX1-RUNX1T1+ AML. Using a highly sensitive RNA-based qRT-PCR assay with a sensitivity of up to 10−6, we identified the prognostic impact of RUNX1-RUNX1T1 TLs and their reduction, and we defined clinically relevant MRD time points during and after therapy that allowed for the identification of patients with a high risk of relapse.
Our study showed that both reduction of RUNX1-RUNX1T1 TLs and achievement of MRD− at defined time points are of significant prognostic importance. First, achievement of MR2.5 after cycle 1 and achievement of MR3.0 after cycle 2 were significantly associated with a reduced risk of relapse. Second, after completion of therapy, specific MRD cutoffs, <83 RUNX1-RUNX1T1 TL/106B2M copies in BM and <5 RUNX1-RUNX1T1 TL/106B2M copies in PB, and achievement of MRD− were independent, favorable prognostic factors for both relapse risk and OS. Finally, during follow-up, serial qRT-PCR analyses enabled prediction of relapse in 77% of patients exceeding a cutoff value of >150 RUNX1-RUNX1T1 TL/106B2M copies in BM and in 84% of patients exceeding a value of >50 RUNX1-RUNX1T1 TL/106B2M copies in PB, respectively.
Our observation that detectable RUNX1-RUNX1T1 TLs after completion of therapy are of prognostic relevance is in accordance with MRD studies also investigating different targets.11 However, for RUNX1-RUNX1T1+ AML only 2 studies have delineated MRD− as a prognostic marker so far, albeit with some limitations. In both studies MRD− was defined as <.001%. In the study from the French AML Intergroup, achievement of CMR in PB at EOT was associated with lower CIR and superior OS; however, CMR in BM had no impact,8 most likely because of the low number of patients achieving MRD− at this time point (22 [30%] of 74 analyzed patients). In the second study, published in 2018, no prespecified time points for MRD assessment were determined.24
In our study, repetitive BM and/or PB MRD− assessment during follow-up was a reliable predictor of sustained remission. For MRD conversion from MRD− to MRD+ we observed a subsequent relapse rate of 64% in PB samples and 24% in BM samples (Figure 3). These results reflect the clinical challenge regarding the interpretation of intermittent MRD+ during follow-up in RUNX1-RUNX1T1+ AML and underscores the indispensability of discriminating molecular persistence at low copy numbers, molecular progression, and molecular relapse on a second sample (after 4 weeks), as recommended by the ELN MRD Working Party11 before diagnosing molecular relapse and considering further therapeutic intervention. For a more precise prediction of relapse risk we aimed to identify specific cutoff values for TLs in BM and PB for patients with at least 1 MRD+ sample. Our analysis revealed that transcript copies >150 in BM and/or >50 in PB were associated with a high rate of subsequent hematologic relapse within a very short period (Figure 3). These cutoff values are lower than those described in the Medical Research Council trial (500 and 100 copies for BM and PB, respectively),10 differences that may be related to the material that was used and the sensitivity of the MRD assays, but also to differences in treatment. Thus, cutoff values are not directly transferable19,20 and have to be validated first. The relapse prediction and median times from exceeding the cutoff values to relapse are hampered by the sampling intervals, which were heterogeneous in our follow-up cohort (Figure 4A). However, virtually all relapses in RUNX1-RUNX1T1+ AML occurred within 1 year after EOT (Figure 3; supplemental Figure 1), which is characteristic of the relapse kinetics of core-binding factor AML.9,10,17,25 Thus, for an earlier and more precise prediction of relapse, MRD assessment should be performed in short (eg, monthly) intervals in the early follow-up period of patients with RUNX1-RUNX1T1+ AML, in order to assess MRD kinetics and to distinguish MRD persistence, molecular progression, and molecular relapse, as defined by the ELN MRD Working Party.11 An important practical issue addressed in our study was whether PB samples could be used for MRD monitoring, in particular in the follow-up period. During therapy, in 44% of the negative PB samples, the corresponding BM samples were positive, whereas during follow-up, the majority (72%) of the paired samples were concordant. Based on our data, we refined the practical guidelines for MRD assessment in RUNX1-RUNX1T1+ AML: (1) along with the current ELN MRD recommendations, BM and PB should be analyzed after each treatment cycle; and (2) during the follow-up period, in particular the first year after EOT, MRD monitoring of PB should be performed monthly, and in patients with TLs exceeding 50 in PB, an increase of MRD >1-log, and/or conversion from MRD− to MRD+, a complementary BM sample should be analyzed promptly (Figure 5).

Proposed refined recommendation based on MRD at EOT and during follow-up. Assessment was conducted in patients with RUNX1-RUNX1T1+ AML, according to the ELN MRD Working Party,11 taking into account MRD status at EOT and during follow-up. Ind, induction therapy; Cons, consolidation therapy.
Proposed refined recommendation based on MRD at EOT and during follow-up. Assessment was conducted in patients with RUNX1-RUNX1T1+ AML, according to the ELN MRD Working Party,11 taking into account MRD status at EOT and during follow-up. Ind, induction therapy; Cons, consolidation therapy.
Prognostic discrimination at earlier time points is still challenging, and current data are controversial. Impact on outcome has been described for MR3.0 after the first treatment cycle10 as well as after the second cycle.9 We confirmed MR3.0 after cycle 2 as a prognostic factor for lower CIR which is in line with the study from the French AML Intergroup that reported ≥3-log BM MRD reduction between diagnosis and the second consolidation cycle to be associated with lower relapse risk and longer RFS.9 Notably, the first prognostic landmark delineated in our study was MR2.5 after cycle 1, which was associated with lower CIR and a trend toward superior OS. The finding that MRD− did not allow earlier risk stratification is probably related to the high sensitivity of our qRT-PCR assay, which is also reflected by the low number of patients becoming MRD− after the first and second treatment cycles.
In line with previous studies,26,27 KITmut was a considerably significant variable at diagnosis, predicting a lower CR rate and inferior outcome, but its prognostic impact was outweighed by MRD status during treatment. In our study, reduction of RUNX1-RUNX1T1 TLs correlated with KITmut, in that KITWT patients achieved significantly deeper MRD reductions and exhibited a significantly higher rate of MRD− at EOT.9,25 However, in multivariable analyses, RUNX1-RUNX1T1 TL and MRD− at EOT were the most considerable variables, whereas KITmut remained a significant cofactor for inferior survival in only some of these models.
In summary, RUNX1-RUNX1T1 MRD monitoring allows for the discrimination of patients at high and low risk of relapse. Achievement of MRD− in both BM and PB, after completion of therapy was the most valuable independent favorable prognostic factor for relapse risk and OS. During follow-up, serial qRT-PCR analyses allowed the delineation of defined cutoff values predicting relapse. Moreover, considering that virtually all relapses occurred within 1 year after EOT with a very short latency from molecular to morphologic relapse, MRD assessment at short intervals during this period is indispensable. Based on our findings we propose a refined practical guidance for molecular MRD monitoring in RUNX1-RUNX1T1+ AML.
Data related to this study may be obtained by e-mail request to the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Agnes Gambietz and Axel Benner (Division of Biostatistics, German Cancer Research Center, Heidelberg, Germany) for determining the cutoff values for prognostic factors in the survival data, with competing risks based on maximally selected log-rank statistics, and the members of the German-Austrian AML Study Group (AMLSG) for providing patient samples and clinical information.
This study was supported in part by the Deutsche Forschungsgemeinschaft (DFG, SFB 1074, project A3 and B3). M.A. was supported by the Else Kröner-Forschungskolleg and the Deutsche Forschungsgemeinschaft (DFG, AG 252/1-1).
Authorship
Contribution: F.G.R., M.A., A.C., H.D., and K.D. designed the study and wrote the manuscript; F.G.R., M.A., A.C., S.K.-S., V.I.G., and N.J. performed the experiments; F.G.R., M.A., A.C., D.W., P.P., H.D., and K.D. analyzed the results; F.G.R. and D.W. performed statistical analyses; F.G.R., M.A., A.C., S.K.-S., V.I.G., N.J., T.S., M.W., M.L., E.K., T.K., K.G., M.R., J.W., W.F., H.A.H., R.G., R.S., K.M., T.H., J.K., R.F.S., F.T., M.H., A.G., L.B., P.P., H.D., and K.D. provided patient samples and clinical information.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A complete list of the members of the German-Austrian Acute Myeloid Leukemia Study Group appears in the supplemental appendix.
Correspondence: Konstanze Döhner, Department of Internal Medicine III, University Hospital of Ulm, Albert-Einstein-Allee 23, D-89081 Ulm, Germany; e-mail: konstanze.doehner@uniklinik-ulm.de.