

Key Points
Defective proresolving response to acute vaso-occlusive events characterizes murine SCD.
Treatment with 17R-RvD1 reduces inflammation and vascular dysfunction in SCD mice.
Abstract
Resolvins (Rvs), endogenous lipid mediators, play a key role in the resolution of inflammation. Sickle cell disease (SCD), a genetic disorder of hemoglobin, is characterized by inflammatory and vaso-occlusive pathologies. We document altered proresolving events following hypoxia/reperfusion in humanized SCD mice. We demonstrate novel protective actions of 17R-resolvin D1 (17R-RvD1; 7S, 8R, 17R-trihydroxy-4Z, 9E, 11E, 13Z, 15E, 19Z-docosahexaenoic acid) in reducing ex vivo human SCD blood leukocyte recruitment by microvascular endothelial cells and in vivo neutrophil adhesion and transmigration. In SCD mice exposed to hypoxia/reoxygenation, oral administration of 17R-RvD1 reduces systemic/local inflammation and vascular dysfunction in lung and kidney. The mechanism of action of 17R-RvD1 involves (1) enhancement of SCD erythrocytes and polymorphonuclear leukocyte efferocytosis, (2) blunting of NF-κB activation, and (3) a reduction in inflammatory cytokines, vascular activation markers, and E-selectin expression. Thus, 17R-RvD1 might represent a new therapeutic strategy for the inflammatory vasculopathy of SCD.
Introduction
Sickle cell disease (SCD) is a genetic disorder of hemoglobin characterized by hemolytic anemia and vaso-occlusive and inflammatory pathologies affecting target organs like lung, brain, bone, and kidney. The disease is characterized by acute and chronic pain, most often in the context of vaso-occlusive crises (VOCs), a definition based on the notion that occlusion of small vessels and/or capillaries by sickled cells is the triggering mechanism for the generation of inflammation and pain. VOCs are the main cause of hospitalization of young adult patients with SCD and can evolve into life-threatening complications.1-4
Novel therapeutic options focusing on physiological processes promoting resolution of inflammation are of interest for treating acute events and for prevention of SCD-related vasculopathy. The resolution process is actively controlled by the temporal and local production of specialized proresolving lipid mediators (SPMs). These include lipoxins (LXs), resolvins (Rv’s), protectins, and maresins from polyunsaturated fatty acids; these novel mechanisms and their mediators offer new therapeutic opportunities for treating inflammation-related disorders in humans.5 SPMs proved effective in limiting the extent and duration of inflammation in experimental asthma,6 infections,7,8 and pain.9 LXA4, RvD1, and their corresponding “aspirin-triggered” epimers, biosynthesized upon acetylation of cyclooxygenase-2 by aspirin, potently stop excessive neutrophil recruitment10 and prevent inflammation-related organ damage in vitro and in vivo.11,12 Because an amplified inflammatory response plays a key role in VOCs and in SCD-related organ damage, we reasoned that inducers of the resolution phase of inflammation might influence the outcome of acute VOCs and attenuate SCD-related organ damage without immune suppression.7,13,14
In this article, using a humanized mouse model for SCD, we report reduced production of endogenous RvD1 in response to hypoxia/reoxygenation (H/R) stress. We show the benefit of exogenous administration of 17R-resolvin D1 (17R-RvD1) in preventing H/R-induced damage in lung and kidney, which are target organs of SCD. These findings suggest that proresolving events during H/R stress, a critical phase in acute sickle cell–related VOCs, may limit and control the magnitude of the inflammatory response in SCD and possibly ameliorate the outcome of VOCs.
Methods
The methods, mouse model, and study design are described in supplemental Methods (available on the Blood Web site).
Results
Specialized proresolving mediators are altered in humanized sickle cell mice
To characterize the bioactive lipid mediator (LM) signature of SCD, targeted liquid chromatography (LC)–tandem mass spectrometry (MS/MS) metabololipidomics was carried out in spleens from healthy control mice (AA) and sickle cell (SS) mice under normoxic conditions or after H/R stress, which is an experimental model of sickle cell–related VOCs. Spleen was chosen because it is one of the target organs of acute VOCs as a result of its very sluggish circulation, which favors HbS polymerization.15 In addition, we recently reported that spleen contributes to the cell inflammatory response and to the generation of specialized proresolving mediators.16,17 As shown in Figure 1, each LM was identified based on LC chromatograms and MS/MS fragmentation, with a minimum of 6 diagnostic ions. We identified LMs from arachidonic acid, eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA) bioactive metabolomes (Tables 1 and 2). In AA mice and SCD mice, the following DHA-derived SPMs were identified: RvD1, 17R-RvD1, RvD5, and RvD6 (but not RvD2, RvD3, or RvD4), as well as protectin D1 and maresin 2, along with their pathway markers. LXA4 was also identified, whereas E-series Rv’s (RvE1-3) were below the limits of detection (∼0.1 pg). Of interest, the amount of SPMs was markedly reduced in SCD mice compared with AA animals under normoxic conditions or exposure to H/R (Tables 1 and 2). Principal component analysis (PCA) data mining was then used to identify distinct LM profiles in SCD mice exposed to hypoxia. PCA showed a clear separation between normoxia and hypoxia SCD samples, with the latter containing higher levels of SPMs (including RvD1, 17R-RvD1, and LXA4), prostaglandin D2 (PGD2), PGE2, PGF2α, and thromboxane B2 (Figure 2A-C), indicating activation of the biosynthesis of select LMs in response to H/R stress. The H/R-induced increase in the biosynthesis of SPMs, as well as prostaglandins, was markedly lower in SCD mice than in AA animals, indicating impairment in specialized proresolving mediators in SCD in response to H/R. To explore the clinical relevance of these findings, we carried out LC-MS/MS metabololipidomics on plasma from human sickle cell subjects in the steady-state. As shown in supplemental Figure 1, we identified significantly increased amounts of PGE2 vs healthy controls. Of interest, specialized proresolving mediators, protectin D1 and RvE1, were also identified in human sickle cell plasma, as well as in normal control plasma, based on matched LC retention time and ≥6 diagnostic MS/MS fragment ions. These data suggest a high proinflammatory pattern in sickle cell patients, supporting the perturbation of proresolving events in human SCD.
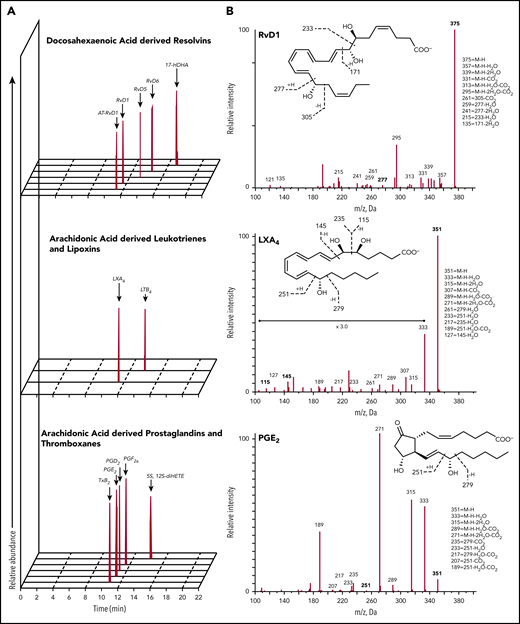
Lipidometabolomic signature of AA and SS mouse spleens. (A) Representative multiple reaction monitoring chromatograms, from spleen samples of sickle cell mice exposed to hypoxia (8% oxygen; 10 hours) and followed by reoxygenation (21% oxygen; 3 hours), used to identify LMs. (B) MS/MS fragmentation spectra used for identification of RvD1, LXA4, and PGE2.
Lipidometabolomic signature of AA and SS mouse spleens. (A) Representative multiple reaction monitoring chromatograms, from spleen samples of sickle cell mice exposed to hypoxia (8% oxygen; 10 hours) and followed by reoxygenation (21% oxygen; 3 hours), used to identify LMs. (B) MS/MS fragmentation spectra used for identification of RvD1, LXA4, and PGE2.
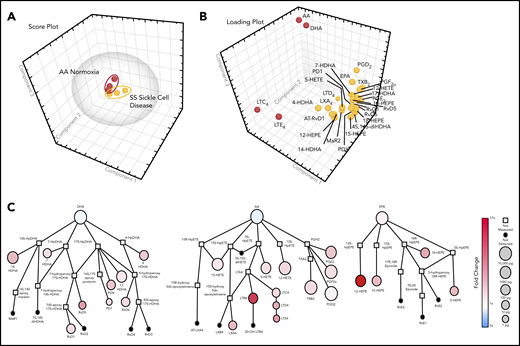
PCA of LMs, specialized proresolving mediator profiles, in healthy and sickle cell mice exposed to H/R stress. (A) Three-dimensional score plot of sickle cell mice under normoxia (red; n = 3) or exposed to 10 hours of hypoxia (8% oxygen) and followed by 18 hours of reoxygenation (yellow; n = 3) for murine spleen samples. Ellipses mark 95% confidence regions. (B) Three-dimensional loading plot. (C) Quantitative pathway network of sickle cell murine spleen samples. Node size represents the mean values (in picograms) of sickle cell normoxia spleen samples (n = 3). Node color denotes the fold changes in sickle cells exposed to hypoxia (10 hours 8%), followed by 18 hours of reoxygenation, vs sickle cell normoxia.
PCA of LMs, specialized proresolving mediator profiles, in healthy and sickle cell mice exposed to H/R stress. (A) Three-dimensional score plot of sickle cell mice under normoxia (red; n = 3) or exposed to 10 hours of hypoxia (8% oxygen) and followed by 18 hours of reoxygenation (yellow; n = 3) for murine spleen samples. Ellipses mark 95% confidence regions. (B) Three-dimensional loading plot. (C) Quantitative pathway network of sickle cell murine spleen samples. Node size represents the mean values (in picograms) of sickle cell normoxia spleen samples (n = 3). Node color denotes the fold changes in sickle cells exposed to hypoxia (10 hours 8%), followed by 18 hours of reoxygenation, vs sickle cell normoxia.
To further characterize D-series Rv biosynthesis and kinetics in humanized SCD mice, ω-3 DHA (C22:6, 1 μg per mouse), as a precursor of D-series Rv, was administered orally to mice from both strains, and the temporal biosynthesis of RvD1 was determined using a competitive immunoenzymatic assay. Because RvD2 was not present in the metabololipidomics profile of SCD mice, we focused on RvD1 (Table 1). The oral route for ω-3 DHA administration was chosen based on our previous studies in mouse models of peritonitis and lung infection,18,19 whereas the time course was chosen to determine the increase in RvD1 plasma levels and return-to-baseline concentrations. To assess possible interference of matrix components with the assay, synthetic RvD1 (40 and 100 pg/mL) was spiked in mouse plasma, and its concentration was measured (supplemental Figure 2A).
As shown in Figure 3A, plasma values of RvD1 did not change significantly in SS mice after DHA administration, whereas they increased markedly in healthy controls, as expected.
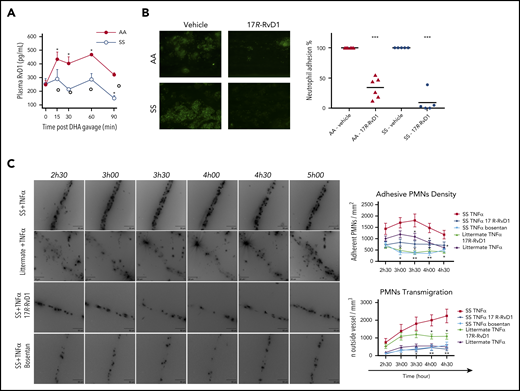
RvD1 reduces ex vivo human neutrophil adhesion and in vivo neutrophil counts in humanized SCD mice, which show decreases in plasma RvD1 values after DHA administration. (A) Kinetics of DHA conversion to proresolving mediator RvD1 following oral administration in AA and SS mice. Levels of RvD1 were determined, using a competitive enzyme immunoassay, in plasma collected from AA and SS mice at the indicated times following DHA gavage. Data are mean ± SD (n = 3). *P < .05 vs baseline for AA mice. (B) Adhesion of neutrophils (green) to TNF-α–activated human microvascular endothelial cell line (HMEC). Blood samples from a healthy donor (AA) and an SCD patient (SS) were incubated for 10 minutes with vehicle or 17R-RvD1 (1 μM) (left panels). A second dose of vehicle or 17R-RvD1 (1 μM) was added before perfusing blood for 40 minutes (at 1 dyn/cm2) through the biochip channels containing activated HMEC-1 monolayers. Percentage of neutrophil adhesion to TNF-α–activated HMECs from AA (n = 6) and SS (n = 5) blood samples incubated with vehicle or 17R-RvD1 (right panel). ***P < .001 vs the corresponding vehicle group. (C) Representative images showing reduced neutrophil recruitment after 17R-RvD1 administration in sickle cell mice (left panels). All of these experiments suggest that 17R-RvD1 influences neutrophil recruitment in SS mice, especially in the context of sustained and intense neutrophil recruitment specific to experimental SCD. Neutrophil adhesion density, defined as the number of adherent neutrophils per square millimeter of endothelial surface, in TNF-α–inflamed venules after 17R-RvD1 treatment in SS mice (SS TNF-α 17R-RvD1) or after dual endothelin receptor antagonism (SS TNF-α bosentan) (upper right panel). Data are mean ± standard error of the mean. P < .001 for SS mice treated with TNF-α and 17-RvD1 vs with TNF-α and vehicle alone (SS TNF-α 17R-RvD1), 2-way analysis of variance (ANOVA). *P < .05, **P < .01 vs vehicle-treated SS with TNF-α, 2-way ANOVA followed by the Tukey multiple-comparison test. Extravascular volume in inflamed venules after 17R-RvD1 or bosentan vs vehicle administration in SS mice (lower right panel). Emigrated neutrophils were visualized and quantified by optical sectioning and 2-dimensional maximum intensity projection. Twenty venules (5 mice) were analyzed in each group. Data are mean ± standard error of the mean. P < .001 for SS mice treated with TNF-α and 17R-RvD1 vs with TNF-α and vehicle alone, 2-way ANOVA. Data for heterozygous AS littermates are provided as control reference for neutrophil adhesion and emigration to tissue after TNF-α. *P < .05, **P < .01 vs vehicle-treated SS with TNF-α, 2-way ANOVA, followed by the Tukey multiple-comparison test.
RvD1 reduces ex vivo human neutrophil adhesion and in vivo neutrophil counts in humanized SCD mice, which show decreases in plasma RvD1 values after DHA administration. (A) Kinetics of DHA conversion to proresolving mediator RvD1 following oral administration in AA and SS mice. Levels of RvD1 were determined, using a competitive enzyme immunoassay, in plasma collected from AA and SS mice at the indicated times following DHA gavage. Data are mean ± SD (n = 3). *P < .05 vs baseline for AA mice. (B) Adhesion of neutrophils (green) to TNF-α–activated human microvascular endothelial cell line (HMEC). Blood samples from a healthy donor (AA) and an SCD patient (SS) were incubated for 10 minutes with vehicle or 17R-RvD1 (1 μM) (left panels). A second dose of vehicle or 17R-RvD1 (1 μM) was added before perfusing blood for 40 minutes (at 1 dyn/cm2) through the biochip channels containing activated HMEC-1 monolayers. Percentage of neutrophil adhesion to TNF-α–activated HMECs from AA (n = 6) and SS (n = 5) blood samples incubated with vehicle or 17R-RvD1 (right panel). ***P < .001 vs the corresponding vehicle group. (C) Representative images showing reduced neutrophil recruitment after 17R-RvD1 administration in sickle cell mice (left panels). All of these experiments suggest that 17R-RvD1 influences neutrophil recruitment in SS mice, especially in the context of sustained and intense neutrophil recruitment specific to experimental SCD. Neutrophil adhesion density, defined as the number of adherent neutrophils per square millimeter of endothelial surface, in TNF-α–inflamed venules after 17R-RvD1 treatment in SS mice (SS TNF-α 17R-RvD1) or after dual endothelin receptor antagonism (SS TNF-α bosentan) (upper right panel). Data are mean ± standard error of the mean. P < .001 for SS mice treated with TNF-α and 17-RvD1 vs with TNF-α and vehicle alone (SS TNF-α 17R-RvD1), 2-way analysis of variance (ANOVA). *P < .05, **P < .01 vs vehicle-treated SS with TNF-α, 2-way ANOVA followed by the Tukey multiple-comparison test. Extravascular volume in inflamed venules after 17R-RvD1 or bosentan vs vehicle administration in SS mice (lower right panel). Emigrated neutrophils were visualized and quantified by optical sectioning and 2-dimensional maximum intensity projection. Twenty venules (5 mice) were analyzed in each group. Data are mean ± standard error of the mean. P < .001 for SS mice treated with TNF-α and 17R-RvD1 vs with TNF-α and vehicle alone, 2-way ANOVA. Data for heterozygous AS littermates are provided as control reference for neutrophil adhesion and emigration to tissue after TNF-α. *P < .05, **P < .01 vs vehicle-treated SS with TNF-α, 2-way ANOVA, followed by the Tukey multiple-comparison test.
We also explored possible abnormalities in the expression of enzymes related to RvD1 and RvD2 biosynthesis that might contribute to the differences in LMs between mouse strains. These were evaluated in lung, as a target organ of SCD, and isolated aorta. No significant difference in messenger RNA levels of COX-2, 5-lipoxygenase, or 12/15-lipoxygenase was present between AA and SS mice (supplemental Figure 2B). We then compared the expression of RvD1 and RvD2 receptors in lungs and aorta from AA and SS mice. Interestingly, ALX/FPR2 expression was downregulated in lung, but not aorta, from SS mice, whereas DRV2/GPR18 messenger RNA levels in lung and aorta were similar in both strains (supplemental Figure 2B). Overall, these results provide the first LM signature of humanized SCD mice and indicate altered proresolving mechanisms in SCD mice.
17R-RvD1 inhibits the ex vivo adhesion of human leukocytes to TNF-α– activated endothelial cells and in vivo in inflamed cremasteric venules from humanized SCD mice
We examined whether proresolving mediators RvD1 (7S, 8R, 17S-trihydroxy-4Z, 9E, 11E, 13Z, 15E, 19Z-docosahexaenoic acid), 17R-RvD1 (7S, 8R, 17R-trihydroxy-4Z, 9E, 11E, 13Z, 15E, 19Z-docosahexaenoic acid), and RvD2 (7S, 16R, 17S-trihydroxy-4Z, 8E, 10Z, 12E, 14E, 19Z-docosahexaenoic acid) might affect the ex vivo adhesion of leukocytes to tumor necrosis factor-α (TNF-α)–activated endothelial cells using a microfluidic chamber. 17R-RvD1 was also tested, because it is known to be longer acting than RvD1 by resisting local enzymatic inactivation.20 In our model, we found that RvD1 and 17R-RvD1 reduced the adhesion of leukocytes from healthy donors and SCD patients to the vascular endothelial surface (Figure 3B), whereas RvD2 was more effective in preventing leukocyte adhesion from AA individuals than from SS individuals (supplemental Figure 3A-B). This agrees with previous evidence in murine leukocytes20 and supports the direct action of 17R-RvD1 on neutrophil adhesion to activated endothelial cells.
As a proof of concept, we evaluated the impact of 17R-RvD1 on neutrophil adhesion and transmigration in TNF-α–mediated inflamed cremasteric microcirculation (Figure 3C).21 In SS mice, the inflammatory challenge with TNF-α induced a marked increase in neutrophil adhesion compared with littermates. The adhesion remained elevated and stable for 3 hours and 30 minutes and then decreased slightly. At the end of the experiment, adhesion density in SS mice challenged with TNF-α was still increased compared with that of littermates (P < .05). 17R-RvD1 markedly inhibited the adhesion of neutrophils in SS mice throughout the time course of the study, with the degree of adhesion being less than that observed in littermates and twofold less than in vehicle-treated SS mice (P < .001) (Figure 3C; supplemental Videos 1-4). Because bosentan, a specific inhibitor of endothelin-1 (ET-1) receptor A-B, prevented neutrophil adhesion in SCD,21,22 we used it as a reference molecule for the effects of 17R-RvD1 (Figure 3C). We found that adhesion efficiency was markedly and similarly decreased in SS mice treated with 17R-RvD1 or bosentan compared with vehicle-treated animals (P < .01; supplemental Figure 3C).
In SCD mice, 17R-RvD1 treatment also significantly decreased neutrophil transmigration in TNF-α–induced inflamed venules (Figure 3C, lower right panel). TNF-α–induced transmigration of neutrophils was significant in both groups of mice compared with vehicle-treated animals (data not shown), but distinct kinetics were observed in SS mice and in littermates. Although neutrophil emigration reached a plateau at 3 hours in the cremasteric microcirculation of littermates, sickle SS mice experienced a sustained and more intense increase in emigration that was double that measured in littermates after 4 hours and 30 minutes (Figure 3C, lower right panel). In SS mice, 17R-RvD1 administration significantly prevented transendothelial migration of neutrophils to tissues, especially at late points (17R-RvD1: ∼10-fold decrease at 2 hours and 30 minutes [P < .01, vs SS mice treated with TNF-α only] and an approximately sevenfold decrease at 4 hours and 30 minutes [P < .01, vs TNF-α–treated mice] [Figure 3C, lower right panel; supplemental Figure 3D; supplemental Video 1]).
17R-RvD1 enhances sickle red cell and polymorphonuclear leukocyte efferocytosis by macrophages
Earlier studies have shown that a defining action of SPMs in the resolution of inflammation is the enhancement of macrophage (MΦ)-mediated phagocytosis of damaged/dead cells.5 Thus, we tested whether 17R-RvD1 may affect neutrophil and red blood cell (RBC) efferocytosis. As shown in Figure 4A-B, treatment of spleen-derived MΦs with 17R-RvD1 resulted in a dose-dependent increase in efferocytosis of RBCs and polymorphonuclear leukocytes (PMNs). Erythrophagocytosis was significantly higher in SS MΦs for SS RBCs compared with AA MΦs for AA erythrocytes at baseline and after 17R-RvD1 (Figure 4A), while no major differences were observed in cross-experiments, namely SS MΦs fed AA RBCs or AA MΦs given SS RBCs (supplemental Figure 4A-B). This might be related to increased phosphatidylserine exposure on SS erythrocytes, favoring erythrophagocytosis23,24 (supplemental Figure 4C), as well as to higher numbers of MΦs in spleens and peritoneal cavity of SS mice compared with wild-type littermates (supplemental Figure 4D). Of note, this was associated with activation of SS MΦs, as indicated by the increased expression of the phosphatidylserine receptors Tim4, CD206, and CD36 that mediate recognition and engulfment of apoptotic cells25,26 (supplemental Figure 4E).
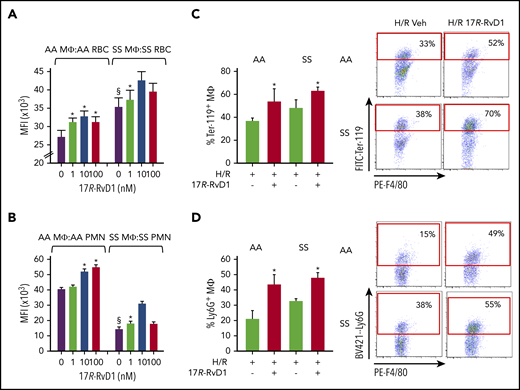
17R-RvD1 enhances phagocytosis of sickle RBCs and PMNs by spleen macrophages. In vitro phagocytosis of carboxyfluorescein diacetate succinimidyl ester–labeled aged RBCs (at 4°C, 18 h; A) or PMNs (37°C, 18 h; B) from AA and SS mice by spleen MΦs (F4/80+ cells), as assessed using flow cytometry. Data are mean ± standard deviation from 6 independent experiments. (C-D) Phagocytosis of RBCs and PMNs by spleen MΦs in vivo in vehicle- or 17R-RvD1–treated AA and SS mice undergoing H/R (left panels). Percentages of MΦs engulfing RBCs (F4/80+ Ter-119+; C) or PMNs (F4/80+ Ly6G+; D) were calculated using flow cytometry. Results are mean ± standard deviation from 3 mice per group. Representative flow cytometry dot plots and gate criteria (right panels). §P < .05 vs AA MΦs + vehicle, *P < .05 vs vehicle-treated cells. MFI, mean fluorescence intensity.
17R-RvD1 enhances phagocytosis of sickle RBCs and PMNs by spleen macrophages. In vitro phagocytosis of carboxyfluorescein diacetate succinimidyl ester–labeled aged RBCs (at 4°C, 18 h; A) or PMNs (37°C, 18 h; B) from AA and SS mice by spleen MΦs (F4/80+ cells), as assessed using flow cytometry. Data are mean ± standard deviation from 6 independent experiments. (C-D) Phagocytosis of RBCs and PMNs by spleen MΦs in vivo in vehicle- or 17R-RvD1–treated AA and SS mice undergoing H/R (left panels). Percentages of MΦs engulfing RBCs (F4/80+ Ter-119+; C) or PMNs (F4/80+ Ly6G+; D) were calculated using flow cytometry. Results are mean ± standard deviation from 3 mice per group. Representative flow cytometry dot plots and gate criteria (right panels). §P < .05 vs AA MΦs + vehicle, *P < .05 vs vehicle-treated cells. MFI, mean fluorescence intensity.
Efferocytosis of SS PMNs was markedly impaired compared with AA, further supporting a defect in proresolution mechanisms in SCD that might be rescued by 17R-RvD1 (Figure 4A-B). Similar results were also obtained when sickle RBCs or PMNs were coincubated with peritoneal MΦs treated with 17R-RvD1 (supplemental Figure 4). We also assessed whether 17R-RvD1 treatment regulates the clearance of PMNs and RBCs in spleen from mice exposed to H/R stress. 17R-RvD1–treated AA and SS mice showed significantly increased splenic MΦs with ingested PMNs (F4/80+ Ly6G+) and RBCs (F4/80+ Ter-119+) compared with vehicle-treated animals, indicating an in vivo enhancement of phagocytosis in 17R-RvD1–treated mice (Figure 4C-D).
17R-RvD1 reduces in vivo neutrophil blood counts during H/R stress in humanized SCD mice
17R-RvD1 significantly reduced the H/R-induced increase in neutrophil counts in both mouse strains (supplemental Figure 5A). This was associated with a decrease in plasma C-reactive protein values in both mouse strains exposed to H/R stress (AA H/R vehicle vs 17R-RvD1, 17.5 ± 0.4 mg/dL vs 10.2 ± 0.9 mg/dL, P < .05; SS H/R vehicle vs 17R-RvD1, 58.2 ± 1.8 mg/dL vs 22.6 ± 3.5 mg/dL, P < .05). Our data support the notion that 17R-RvD diminishes the H/R systemic inflammation in SS mice.
17R-RvD1 reduces lung injury and prevents the activation of inflammatory-related transcriptional factors
In AA and SS mice, 17R-RvD1 reduced bronchoalveolar lavage liquid (BAL) protein content and leukocyte counts (Figure 5A). This was associated with a significant decrease in lung inflammatory cell infiltrates, as well as in mucus and thrombi formation (Table 3; supplemental Figure 5B-C).28,29 Consistent with these observations, we found a significant decrease in the active forms of NF-κB and Nrf2 induced by H/R in AA and SS mice treated with 17R-RvD1 (Figure 5B). Because we previously reported that Rv’s reduce NF-κB activation by modulation of microRNAs (miRNAs),30 we assessed miRNAs known to be involved in acute inflammation (ie, miR-126, miR-181b, and miR-146b). miR-126 was reduced during H/R compared with during normoxia in both mouse strains (Figure 5C; supplemental Figure 5D). 17R-RvD1 significantly enhanced miR-126 in SS mice compared with vehicle-treated H/R SS mice, whereas no significant changes were observed in miR-181b and miR-146b expression in AA and SS mice (supplemental Figure 5D). These findings suggest a specific regulatory effect of 17R-RvD1 on miR-126 in SS mice exposed to H/R stress. In AA mice exposed to H/R, no major changes in NF-κB activation or miR-126 levels were observed in vehicle-treated animals, which might be related to the duration of H/R. This may also explain the lack of effect of 17R-RvD1 on NF-κB and miR-126 in AA mice. Collectively, our data indicate that 17R-RvD1 prevents the H/R-induced amplified inflammatory response in lung from SS mice by targeting specific molecular pathways (ie, NF-κB signaling, redox responses, and miRNA-mediated regulation of gene expression).
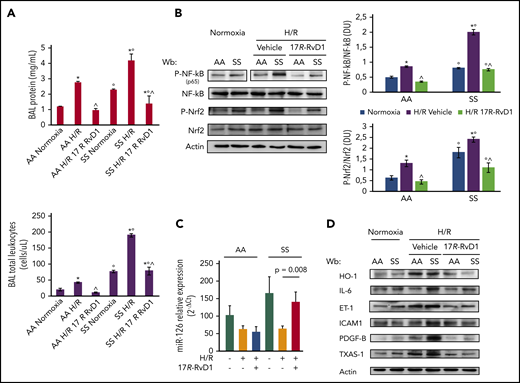
17R-RvD1 prevents H/R activation of acute inflammatory response pathways and reduces SCD vascular vulnerability through multimodal action on NF-κB activation. (A) BAL protein content (upper panel) and leukocyte content (lower panel) from AA and SS mice under normoxia and treated with vehicle or 17R-RvD1 (100 ng) and exposed to H/R: hypoxia (8% oxygen; 10 hours), followed by reoxygenation (21% oxygen; 3 hours) (upper panel). All data are mean ± SD (n = 6). (B) Immunoblot analysis, using specific antibodies against phosphorylated (P-)NF-κB, NF-κB, P-Nrf2, and Nrf2, in lung from AA and SS mice treated as in (A) (left panel). Vertical line(s) in NF-κB, P-p65 gel have been inserted to indicate a repositioned gel lane. One representative gel from 6 gels with similar results is presented. Densitometric analysis of immunoblots (right panels). Data are mean ± SD (n = 6 in each group). (C) Expression of miR-126 (mmu–miR-126-5p), as determined using quantitative polymerase chain reaction, in the lungs of AA and SS mice undergoing H/R and 17R-RvD1 treatment. Results are mean ± SD from 3 to 6 mice per group. (D) Immunoblot analysis, using specific antibodies against HO-1, IL-6, ET-1, ICAM-1, PDGF-B, and TXAS-1, of lung from AA and SS mice treated as in (B). One representative gel from 6 gels with similar results is shown. Densitometric analysis immunoblots are shown in supplemental Figure 2A. *P < .05 vs normoxia, °P < .05 vs healthy mice (AA), ^P < .05 vs vehicle.
17R-RvD1 prevents H/R activation of acute inflammatory response pathways and reduces SCD vascular vulnerability through multimodal action on NF-κB activation. (A) BAL protein content (upper panel) and leukocyte content (lower panel) from AA and SS mice under normoxia and treated with vehicle or 17R-RvD1 (100 ng) and exposed to H/R: hypoxia (8% oxygen; 10 hours), followed by reoxygenation (21% oxygen; 3 hours) (upper panel). All data are mean ± SD (n = 6). (B) Immunoblot analysis, using specific antibodies against phosphorylated (P-)NF-κB, NF-κB, P-Nrf2, and Nrf2, in lung from AA and SS mice treated as in (A) (left panel). Vertical line(s) in NF-κB, P-p65 gel have been inserted to indicate a repositioned gel lane. One representative gel from 6 gels with similar results is presented. Densitometric analysis of immunoblots (right panels). Data are mean ± SD (n = 6 in each group). (C) Expression of miR-126 (mmu–miR-126-5p), as determined using quantitative polymerase chain reaction, in the lungs of AA and SS mice undergoing H/R and 17R-RvD1 treatment. Results are mean ± SD from 3 to 6 mice per group. (D) Immunoblot analysis, using specific antibodies against HO-1, IL-6, ET-1, ICAM-1, PDGF-B, and TXAS-1, of lung from AA and SS mice treated as in (B). One representative gel from 6 gels with similar results is shown. Densitometric analysis immunoblots are shown in supplemental Figure 2A. *P < .05 vs normoxia, °P < .05 vs healthy mice (AA), ^P < .05 vs vehicle.
17R-RvD1 resolves hypoxia-induced lung inflammatory vasculopathy and modulates pulmonary extravascular remodeling
To assess whether 17R-RvD1 was protective against H/R-induced oxidation and vascular dysfunction, we evaluated lung expression of heme-oxygenase-1 (HO-1), a cytoprotective system related to Nrf2,31-33 proinflammatory cytokines,22,34 and markers of vascular endothelial activation and neutrophil vascular recruitment.34-36 As shown in Figure 5D, we found a significant reduction in HO-1 lung expression, supporting an antioxidant effect of 17R-RvD1 during H/R stress in both mouse strains (supplemental Figure 5E). Because previous studies have shown upregulation of HO-1 by RvD1 treatment,37 we evaluated HO-1 expression in both mouse strains exposed to hypoxia, followed by 18 hours of reoxygenation, and treated with 17R-RvD1 at the end of hypoxia. In AA mice, we observed increased HO-1 expression in posthypoxia 17R-RvD1–treated mice, whereas, again, HO-1 was downregulated in SS mice (supplemental Figure 6A). This finding indicates that the difference in HO-1 behavior in response to 17R-RvD1 is related to the SCD phenotype, which is characterized by chronic inflammation with persistent activation of Nrf2.2,34,38,39
We then evaluated the key proinflammatory cytokines interleukin-1β (IL-1β) and IL-6, which were reduced significantly in cell-free lung homogenates from AA and SS mice treated with 17R-RvD1 (supplemental Figure 6B). Lung IL-6 and ET-1 expression was significantly lower in 17R-RvD1–treated mice exposed to H/R than in vehicle-treated groups (Figure 5D; supplemental Figure 6B). A similar effect was also observed in lungs from SS mice exposed to hypoxia followed by 18 hours of reoxygenation, when treated with 17R-RvD1 at the end of the hypoxia period (supplemental Figure 6C).
The observed reduction in ET-1 values in H/R-exposed SS mice treated with 17R-RvD1 is relevant, because ET-1 plays a pivotal role as a proinflammatory cytokine and a potent vasoconstrictive molecule, further worsening local vascular dysfunction related to SCD.22 In support of this idea, we found a significant reduction in H/R-induced intracellular adhesion molecule-1 (ICAM-1) expression in SS mice, whereas no major differences were observed in AA mice (Figure 5D; supplemental Figure 5E). We also found a reduction in E-selectin expression in SS mice exposed to H/R and treated with 17R-RvD1 compared with vehicle-treated SS animals (supplemental Figure 6C).
We then evaluated the expression of platelet-derived growth factor-B (PDGF-B), which is known to be a factor involved in lung extravascular matrix remodeling in response to hypoxia.35,36 As shown in Figure 5D, 17R-RvD1 prevented the H/R-induced increased expression of PDGF-B in both mouse models (supplemental Figure 5E).
Finally, we analyzed the expression of thromboxane-synthase (TXAS-1), which is placed under the control of NF-κB and Nrf2.40-42 TXAS-1 has been linked to the activation of vascular endothelial cells and platelets in other models of ischemia-reperfusion damage.41,42 In both mouse models, 17R-RvD1 markedly reduced the H/R upregulation of TXAS-1 compared with vehicle-treated animals (Figure 5D; supplemental Figure 5E). Overall, these studies provide in vivo evidence that 17R-RvD1 treatment protects SS mice against H/R-induced lung injury by suppressing key mediators of inflammation and vascular damage.
Proresolving action of 17R-RvD1 diminishes H/R-induced kidney damage in SS mice
We also examined whether 17R-RvD1 may be protective against H/R-induced kidney injury in SS mice.43-45 We found that, in SS mice, treatment with 17R-RvD1 significantly prevented the H/R-induced increase in creatinine and blood urea nitrogen plasma values compared with vehicle treatment (Figure 6A). Histopathologic analysis of kidneys from control and SS mice exposed to H/R and treated with 17R-RvD1 revealed a marked reduction in glomerular inflammatory cell infiltration, glomerular sclerosis, and thrombi formation compared with vehicle-treated H/R animals (Figure 6B-F; Table 3).22,46 In agreement, we found a reduction in H/R-induced activation of NF-κB in kidney from both mouse strains (Figure 6G; supplemental Figure 6D). Of note, Nrf2 was activated only in SS mice in response to H/R stress, and it was reduced significantly by 17R-RvD1 administration (Figure 6G; supplemental Figure 6D). We found a reduction in HO-1 expression in H/R SS mice treated with 17R-RvD1 compared with vehicle-treated SS animals (Figure 6H; supplemental Figure 7A), whereas HO-1 was undetectable in healthy mice under normoxic conditions or after exposure to H/R (Figure 6H; supplemental Figure 7A).
![Figure 6. 17R-RvD1 protects kidney from sickle cell–related acute injury, prevents inflammatory vascular activation, and positively modulates antifibrotic let7c miRNA. (A) Plasma creatinine (upper panel) and blood urea nitrogen (BUN) (lower panel) levels in AA and SS mice under normoxic conditions or treated with vehicle or 17R-RvD1 (100 ng) and exposed to H/R: hypoxia (8% oxygen; 10 hours), followed by reoxygenation (21% oxygen; 3 hours). Data are mean ± SD (n = 6). *P < .05 vs normoxia, °P < .05 vs healthy mice (AA), ^P < .05 vs vehicle. Hematoxylin and eosin–stained sections of kidney tissue from AA and SS mice treated with vehicle [AA (B); SS (D-E)] or 100 ng of 17R-RvD1 [AA (C); SS (F)] exposed to H/R: hypoxia (8% oxygen; 10 hours) followed by reoxygenation (21% oxygen; 3 hours) (original magnification ×400). Sections of kidney from mice given 17R-RvD1 (C,F) show less glomerular inflammatory cellular infiltrate (in the form of lymphocytes, neutrophils, and plasma cells), glomerular sclerosis (arrow), and thrombi (arrow) compared with vehicle-treated mice (B,D-E); also see Table 1). (G) Immunoblot analysis, using specific antibodies against phosphorylated (P-)NF-κB, NF-κB, P-Nrf2, and Nrf2, in kidney from AA and SS mice treated as in (B). One representative gel from 6 gels with similar results is shown. Densitometric analysis of immunoblots is shown in supplemental Figure 2A. (H) Immunoblot analysis, using specific antibodies against HO-1, IL-6, ET-1, VCAM-1, and TXAS-1, of kidney from AA and SS mice treated as in (B). One representative gel from 6 gels with similar results is shown. Vertical line(s) in ET-1 gel have been inserted to indicate a repositioned gel lane. Densitometric analysis immunoblots are shown in supplemental Figure 2A. (I) Effect of 17R-RvD1 on kidney let7c expression. Levels of miRNA let7c were quantified, using real-time polymerase chain reaction, in kidneys collected from AA and SS mice that were treated as above. Results are mean ± SD from 3 to 6 mice per group.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/133/3/10.1182_blood-2018-07-865378/7/m_blood865378f6.png?Expires=1769099784&Signature=mbO~s1KvQGRQzhdxPlC1GUmKRAou7pjFoSQMdcd2hqW0Ru4euCVuujaDL0tR~zrhkR1P2rvllAWP9KsqARzqBg8u~j8nQVhb4~xD4GI11p~qPrlpA0tVUBZ6rYXP4LQU1kITsLmux63IMmxHkAwRCGH1vMiRB9ki5GfFXtbnSICsYFkM3aoVet9KecKcD6zSe-63C43qttSW78MK3oqOdk66ZBocsbKBSCSwm5APOWXCUN1i4nhxBRTcJkFEYC6kYzWC-zr4ZMSMZj-IeWa9fgYBvmKQpBg9VYMBxEQbtNRp5piXJfJsdVSH4E2CRUbKZ5B4dlWctKuD5vB38oJfNw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
17R-RvD1 protects kidney from sickle cell–related acute injury, prevents inflammatory vascular activation, and positively modulates antifibrotic let7c miRNA. (A) Plasma creatinine (upper panel) and blood urea nitrogen (BUN) (lower panel) levels in AA and SS mice under normoxic conditions or treated with vehicle or 17R-RvD1 (100 ng) and exposed to H/R: hypoxia (8% oxygen; 10 hours), followed by reoxygenation (21% oxygen; 3 hours). Data are mean ± SD (n = 6). *P < .05 vs normoxia, °P < .05 vs healthy mice (AA), ^P < .05 vs vehicle. Hematoxylin and eosin–stained sections of kidney tissue from AA and SS mice treated with vehicle [AA (B); SS (D-E)] or 100 ng of 17R-RvD1 [AA (C); SS (F)] exposed to H/R: hypoxia (8% oxygen; 10 hours) followed by reoxygenation (21% oxygen; 3 hours) (original magnification ×400). Sections of kidney from mice given 17R-RvD1 (C,F) show less glomerular inflammatory cellular infiltrate (in the form of lymphocytes, neutrophils, and plasma cells), glomerular sclerosis (arrow), and thrombi (arrow) compared with vehicle-treated mice (B,D-E); also see Table 1). (G) Immunoblot analysis, using specific antibodies against phosphorylated (P-)NF-κB, NF-κB, P-Nrf2, and Nrf2, in kidney from AA and SS mice treated as in (B). One representative gel from 6 gels with similar results is shown. Densitometric analysis of immunoblots is shown in supplemental Figure 2A. (H) Immunoblot analysis, using specific antibodies against HO-1, IL-6, ET-1, VCAM-1, and TXAS-1, of kidney from AA and SS mice treated as in (B). One representative gel from 6 gels with similar results is shown. Vertical line(s) in ET-1 gel have been inserted to indicate a repositioned gel lane. Densitometric analysis immunoblots are shown in supplemental Figure 2A. (I) Effect of 17R-RvD1 on kidney let7c expression. Levels of miRNA let7c were quantified, using real-time polymerase chain reaction, in kidneys collected from AA and SS mice that were treated as above. Results are mean ± SD from 3 to 6 mice per group.
17R-RvD1 protects kidney from sickle cell–related acute injury, prevents inflammatory vascular activation, and positively modulates antifibrotic let7c miRNA. (A) Plasma creatinine (upper panel) and blood urea nitrogen (BUN) (lower panel) levels in AA and SS mice under normoxic conditions or treated with vehicle or 17R-RvD1 (100 ng) and exposed to H/R: hypoxia (8% oxygen; 10 hours), followed by reoxygenation (21% oxygen; 3 hours). Data are mean ± SD (n = 6). *P < .05 vs normoxia, °P < .05 vs healthy mice (AA), ^P < .05 vs vehicle. Hematoxylin and eosin–stained sections of kidney tissue from AA and SS mice treated with vehicle [AA (B); SS (D-E)] or 100 ng of 17R-RvD1 [AA (C); SS (F)] exposed to H/R: hypoxia (8% oxygen; 10 hours) followed by reoxygenation (21% oxygen; 3 hours) (original magnification ×400). Sections of kidney from mice given 17R-RvD1 (C,F) show less glomerular inflammatory cellular infiltrate (in the form of lymphocytes, neutrophils, and plasma cells), glomerular sclerosis (arrow), and thrombi (arrow) compared with vehicle-treated mice (B,D-E); also see Table 1). (G) Immunoblot analysis, using specific antibodies against phosphorylated (P-)NF-κB, NF-κB, P-Nrf2, and Nrf2, in kidney from AA and SS mice treated as in (B). One representative gel from 6 gels with similar results is shown. Densitometric analysis of immunoblots is shown in supplemental Figure 2A. (H) Immunoblot analysis, using specific antibodies against HO-1, IL-6, ET-1, VCAM-1, and TXAS-1, of kidney from AA and SS mice treated as in (B). One representative gel from 6 gels with similar results is shown. Vertical line(s) in ET-1 gel have been inserted to indicate a repositioned gel lane. Densitometric analysis immunoblots are shown in supplemental Figure 2A. (I) Effect of 17R-RvD1 on kidney let7c expression. Levels of miRNA let7c were quantified, using real-time polymerase chain reaction, in kidneys collected from AA and SS mice that were treated as above. Results are mean ± SD from 3 to 6 mice per group.
The proresolving effects of 17R-RvD1 in SS mice were further supported by the reduction in the H/R-induced increase in IL-6 expression (Figure 6H; supplemental Figure 7A) and ET-1 (Figure 6D; supplemental Figure 7A). ET-1 also increased in AA mice in response to H/R and diminished with administration of 17R-RvD1 (Figure 6D; supplemental Figure 7A).
At baseline, SS mice already showed higher values of HO-1, ET-1, VCAM-1, and TXAS-1, indicating a chronic kidney inflammatory vasculopathy related to the SCD phenotype (Figure 6D; supplemental Figure 7A). Markers of vascular endothelial and platelet activation, such as VCAM-1, TXAS-1, and E-selectin, were increased in H/R-exposed mice and were reduced by treatment with 17R-RvD1 (Figure 6D; supplemental Figure 7A-B).
17R-RvD1 reduces the inflammatory-related profibrotic stimulus induced by acute VOC
To explore the mechanisms associated with the effects of 17R-RvD1 on sickle cell kidney damage, we evaluated the expression of miRNA let7c, which has previously been shown to be targeted by SPMs and to reduce renal fibrosis through the transforming growth factor-β1 system.47 This latter effect seems to be important in sickle cell kidney disease via a partially unknown cascade.43,44 We found that let7c was markedly increased in response to H/R stress in humanized AA mice (Figure 6I). This increase was more pronounced in the SS mouse strain, suggesting the existence of an endogenous circuit aimed at limiting transforming growth factor-β1 signaling and, subsequently, kidney fibrosis (Figure 6I). 17R-RvD1 further enhanced the antifibrotic miRNA let7c in AA and SS animals undergoing H/R (Figure 6I). These data indicate that 17R-RvD1 mitigates the H/R-induced acute kidney damage and resolves the related amplified inflammation and vascular dysfunction, preventing the activation of profibrotic mechanism(s).
17R-RvD1 protects against progression of inflammatory vasculopathy during acute H/R stress in SS mice
Given the beneficial effects of proresolving LM treatment on different models of vascular dysfunction and inflammatory vasculopathy,48-51 we tested the impact of 17R-RvD1 treatment on the inflammatory vasculopathy of SCD. Administration of 17R-RvD1 significantly reduced the H/R-induced expression of HO-1 in isolated aortas from both mouse strains (Figure 7A). In addition, we found a significant reduction in the H/R-induced expression of ET-1 and VCAM-1 compared with vehicle-treated mice (Figure 7A). The beneficial effect of 17R-RvD1 on VCAM-1 expression was further confirmed by immunochemistry (Figure 7B). Aorta from SS mice exposed to H/R showed increased ICAM-1, which was prevented by 17R-RvD1 treatment (Figure 7A-B). Of note, a reduction in H/R-induced VCAM-1 and ICAM-1 was also observed in aorta from SS mice exposed to hypoxia, followed by 18 hours of reoxygenation, and treated with 17R-RvD1 at the end of the hypoxia period (supplemental Figure 7C). These data indicate that resolution of H/R-induced inflammatory vasculopathy might be altered in SCD mice, requiring the administration of exogenous proresolving molecules, such as 17R-RvD1, to prevent severe vascular dysfunction and disease progression.
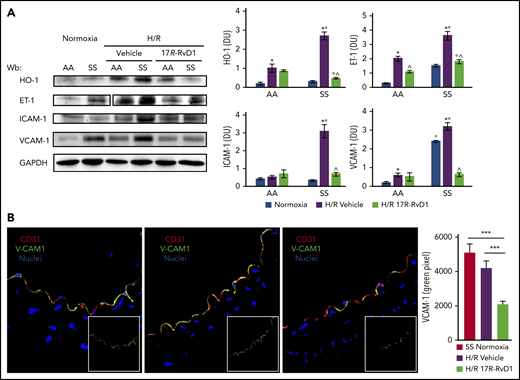
17R-RvD1 reduces vascular vulnerability in SCD mice during acute VOCs. (A) Immunoblot analysis, using specific antibodies against HO-1, IL-6, ET-1, VCAM-1, and TXAS-1, of isolated aorta from AA and SS mice under normoxic conditions and treated with vehicle or 17R-RvD1 (100 ng) and exposed to H/R: hypoxia (8% oxygen; 10 hours) followed by reoxygenation (21% oxygen; 3 hours) (left panel). One representative gel from 6 gels with similar results is shown. Vertical line(s) have been inserted to indicate a repositioned gel lane. Densitometric analysis of immunoblots (right panels). All data are mean ± SD (n = 6). *P < .05 vs normoxia, °P < .05 vs healthy mice (AA), ^P < .05 vs vehicle. (B) VCAM-1 expression was evaluated in aortas isolated from AA or SS mice exposed to H/R and treated with 17R-RvD1, as above (left panels). Following incubation with the indicated antibodies, fluorescence intensities of VCAM-1 staining (green channel) were quantified in digital images (2 to 6 microscopic fields per sample taken at a ×630 magnification scale) by selecting green stained vessels using the Magic Wand Tool in Adobe Photoshop. Semiquantitative analysis of the number of pixels in the selected fields is shown in a bar graph (right panel). *P < .05.
17R-RvD1 reduces vascular vulnerability in SCD mice during acute VOCs. (A) Immunoblot analysis, using specific antibodies against HO-1, IL-6, ET-1, VCAM-1, and TXAS-1, of isolated aorta from AA and SS mice under normoxic conditions and treated with vehicle or 17R-RvD1 (100 ng) and exposed to H/R: hypoxia (8% oxygen; 10 hours) followed by reoxygenation (21% oxygen; 3 hours) (left panel). One representative gel from 6 gels with similar results is shown. Vertical line(s) have been inserted to indicate a repositioned gel lane. Densitometric analysis of immunoblots (right panels). All data are mean ± SD (n = 6). *P < .05 vs normoxia, °P < .05 vs healthy mice (AA), ^P < .05 vs vehicle. (B) VCAM-1 expression was evaluated in aortas isolated from AA or SS mice exposed to H/R and treated with 17R-RvD1, as above (left panels). Following incubation with the indicated antibodies, fluorescence intensities of VCAM-1 staining (green channel) were quantified in digital images (2 to 6 microscopic fields per sample taken at a ×630 magnification scale) by selecting green stained vessels using the Magic Wand Tool in Adobe Photoshop. Semiquantitative analysis of the number of pixels in the selected fields is shown in a bar graph (right panel). *P < .05.
Discussion
For the first time, our data identify altered proresolving processes in humanized sickle cell mice. This is supported by the abnormal LM profile, with a clear defect in SPMs in response to H/R stress in the spleen of SCD mice (Figures 1 and 2; Table 1), reduced ALX expression in SS lungs, and blunted RvD1 biosynthesis post-DHA administration. Thus, the reduction in RvD1 biosynthesis following DHA administration in SCD may most likely be related to a defect in the activation of biosynthetic pathways and enzymes, whose activity may not be strictly related to their expression levels (Figure 3). Moreover, given that no significant differences were found in RvD2 levels following DHA gavage between AA and SS mice, it is unlikely that differences in RvD1 plasma levels arise from abnormalities in the bioavailability of DHA in SCD. This altered response could be further affected by the reported increased amounts of arachidonic acid (the biosynthetic precursor of leukotriene B4) in SCD,34 which could contribute to delayed resolution of inflammation and worsening tissue damage in SS mice exposed to H/R stress. Indeed, intravital microscopy data indicate the important role of 17R-RvD1 in reducing the adhesion of neutrophils to activated endothelium and their transmigration in the presence of inflammatory SCD vasculopathy. This is of particular interest, because neutrophil adhesion in microcirculation is critical for generation of heterothrombi, which represent an early event in acute VOC.52
The enhanced phagocytosis of sickle RBCs in vitro and in vivo by MΦs in spleen from 17R-RvD1–treated mice exposed to H/R stress provides an additional mechanism of action of 17R-RvD1 in accelerating the proresolving events in SCD. This is in agreement with previous reports on enhanced uptake of apoptotic PMNs and lymphocytes by lung MΦs during chronic pulmonary infection53 and of 17R-RvD1 increasing the phagocytic uptake of Escherichia coli.8 Taken together, our data indicate that 17R-RvD1 administration supports the altered resolution of inflammation in SCD by preventing neutrophil recruitment and adhesion to microvascular endothelium, as well as by accelerating the clearance of damaged cells. This conclusion should be tempered by the limitation that we have only demonstrated altered RvD1 function in spleen and peripheral blood. Thus, the assumption that the spleen can be used as a reporter organ for SPMs needs to be validated at the local level.
In SCD mice, restoration of proresolving processes by 17R-RvD1 administration is extremely important in critical setting for SCD, such as H/R stress. We show that administration of 17R-RvD1 reduces the activation of NF-κB and modulates the expression of miR-126 in lung from SS mice exposed to H/R; in parallel, expression of ICAM-1 adhesion molecules on vascular endothelial cells is also significantly reduced, most likely as a consequence of blunted TNF-α/NF-κB activation, which is known to regulate leukocyte recruitment.54 17R-RvD1 also resulted in a significant reduction in proinflammatory cytokines, particularly ET-1, a key proinflammatory and vaso-active cytokine that is extremely important in SCD vasculopathy.21,22
In SS mice, 17R-RvD1 also affected biomarkers associated with H/R-induced kidney damage. In this regard, 17R-RvD1 prevented H/R activation of acute-phase transcriptional factors like NF-κB and Nrf2 in kidney from SS mice. An earlier work by Duffield et al demonstrated that Rv’s reduce inflammation and damage in acute kidney disease.55 In addition, the observation that 17R-RvD1 enhanced let7c expression, interfering with activation of profibrotic mechanisms, further supports the importance of restoring proresolving events in SCD. Together with results on miR-126, these findings indicate the existence of common proresolution circuits activated by different SPMs (ie, 17R-RvD1 and LXA4) involving miRNAs in SCD.
In conclusion, the results of the present study established the presence of an imbalance between proinflammatory and proresolving events in SS mice, at baseline conditions and after exposure to H/R stress mimicking acute VOCs. Mechanistically, we report that RvD1 plays a crucial role in preventing the adhesion of neutrophils to vascular endothelium in SCD and demonstrate that administration of 17R-RvD1 is protective against H/R-induced lung and kidney injury, by acting on NF-κB function and preventing amplified inflammatory response. Thus, SCD mice may be more vulnerable to inflammatory vasculopathy due to altered proresolving processes (supplemental Figure 8). Treatment with 17R-RvD1 restores the inflammatory imbalance and prevents H/R-induced abnormal vascular activation and inflammatory response, preventing H/R organ damage and disease progression. Our results show that 17R-RvD1, as a new therapeutic strategy, will need to be tested in appropriate human trials in SCD. Taken together, our data provide a rationale to develop new therapeutic approaches using proresolving mediators for the clinical management of acute SCD–related events.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported in part by grants from Fondo Universitario Ricerca 2016-2017 (L.D.F.), the Italian Ministry of Health (GR-2011-02349730; A.R.), the Laboratory of Excellence on Red Blood Cells–Globule Rouse Lab of Excellence (P.-L.T.), and the National Institutes of Health, National Institute of General Medical Sciences (grant R01 GM038765; C.N.S.).
Authorship
Contribution: A.M., L.D.F., A.R., and C.B. designed the experiments, analyzed data, and wrote the manuscript; C.N.S. helped in the design of experiments and contributed to manuscript preparation; O.W. carried out the histologic analyses; A.M., E.F., A.S., L.D.F., P.C.N., and I.R.R. carried out the experiments; I.A. and A.I. performed the molecular experiments and analyzed the data; V.B. selected sickle cell patients for in vitro studies; B.K. and W.E.N. carried out ex vivo experiments, analyzed data, and wrote the manuscript; A.L. carried out ex vivo experiments on aorta immunohistochemistry and analyzed data; and P.-L.T. and T.M. carried out ex vivo intravital experiments, analyzed data, and wrote the manuscript
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Carlo Brugnara, Department of Laboratory Medicine, Boston Children’s Hospital, 300 Longwood Ave, Bader 760, Boston, MA 02115; e-mail: carlo.brugnara@childrens.harvard.edu.

