

Key Points
Deficiency of Sirt-1 reduces T-cell pathogenicity in GVHD by enhancing p53 acetylation in T cells and promoting iTreg stability.
Treatment with a Sirt-1 inhibitor reduces Tfh generation, B-cell activation, and plasma cell differentiation during cGVHD development.

Abstract
Graft-versus-host disease (GVHD) remains one of the major complications after allogeneic bone marrow transplantation (allo-BMT). Sirtuin-1 (Sirt-1) plays a crucial role in various biological processes including cellular senescence, metabolism, and inflammatory responses. Sirt-1 deacetylation regulates different transcription factors that are important for modulating immune responses. In the current study, we addressed the role of Sirt-1 in GVHD induction by employing Sirt-1 conditional knockout mice as well as a pharmacological Sirt-1 inhibitor. Using major histocompatibility complex (MHC)–mismatched and MHC-matched murine BMT models, we found that Sirt-1−/− T cells had a reduced ability to induce acute GVHD (aGVHD) via enhanced p53 acetylation. Sirt-1-deficient T cells also promoted induced regulatory T cell (iTreg) differentiation and inhibited interferon-γ production after allo-BMT. Sirt-1 deletion in iTregs increased Foxp3 stability and restrained iTreg conversion into pathogenic T cells. Furthermore, we found that administration with a Sirt-1 inhibitor, Ex-527, significantly improved recipient survival and clinical scores, with no signs of tumor relapse. These results indicate that Sirt-1 inhibition can attenuate GVHD while preserving the graft-versus-leukemia effect. Consistently, Sirt-1-deficient T cells also displayed a remarkably reduced ability to induce chronic GVHD (cGVHD). Mechanistic studies revealed that Sirt-1 deficiency in T cells enhanced splenic B-cell reconstitution and reduced follicular T helper cell development. Sirt-1 deficiency in T cells modulated donor B-cell responses reducing both B-cell activation and plasma cell differentiation. In addition, therapeutic Sirt-1 inhibition could both prevent cGVHD and reduce established cGVHD. In conclusion, Sirt-1 is a promising therapeutic target for the control of aGVHD and cGVHD pathogenesis and possesses high potential for clinical application.
Introduction
Sirtuin-1 (Sirt-1) belongs to the class III histone deacetylase family, which collectively deacetylates a broad range of transcription factors and coregulators, subsequently resulting in up- or downregulation of target gene expression. Sirt-1 requires nicotinamide adenosine dinucleotide as a cosubstrate on deacetylation.1-3 Acetylation/deacetylation is one of the major posttranslational modifications affecting several cellular signaling processes, as well as the metabolism process.4,5 Sirt-1 interacts with several target substrates that have been previously identified, including p53,6-8 Foxo-family members,9,10 AP-1,11 and NF-κb.12 Sirt-1 was demonstrated to regulate cell survival and proliferation via p53 inactivation. Hence, Sirt-1 is recruited by the repressor Mdm2-mediated p53 acetylation. Loss of Sirt-1 leads to hyperacetylation of p53, which prevents its binding to Mdm2, ultimately resulting in cell cycle arrest and apoptosis.6-8 A previous study reported that Sirt-1 negatively regulates T-cell activation through deacetylation of c-Jun and subsequent inactivation of AP-1. Thus, Sirt-1-deficient mice failed to maintain T-cell tolerance and developed severe experimental autoimmune encephalomyelitis (EAE).11 Another study using specific deletion of Sirt-1 in T cells via a Cre-lox system had contradictory results, as Sirt-1 inhibition decreased Th17 differentiation and alleviated disease severity.13 The latter finding was further supported by other studies demonstrating that conditional knockout (KO) of Sirt-1 in T cells promoted induced regulatory T cell (iTreg) differentiation and had enhanced Foxp3 acetylation, thereby prolonging allograft survival.14,15
Graft-versus-host disease (GVHD) remains one of the major complications after allogeneic bone marrow transplantation (allo-BMT). Acute GVHD (aGVHD) is distinguished by uncontrolled activation, migration, and proliferation of allogeneic donor T cells, as well as their production of pro-inflammatory cytokines in GVHD target organs.16 In contrast, chronic GVHD (cGVHD) pathogenesis involves several immune cell types, including pathogenic T- and B-cell interactions and follicular T helper cell (Tfh) generation. Plasma cell differentiation and autoantibody production have also been demonstrated to contribute to disease pathology.17-20
In the current study, we demonstrate that Sirt-1 inhibition, either by genetic ablation or pharmacological blockade, diminished T-cell activation and pathogenicity in GVHD through enhancing p53 acetylation and signaling. Sirt-1 deficiency in T cells not only decreased alloreactivity of donor T cells but also promoted iTreg differentiation after allo-BMT. Furthermore, Sirt-1−/− CD4 iTregs retained Foxp3 expression in inflammatory environments as a result of upregulation of interleukin (IL)-2Rα expression, resulting in increased stability and a reduced conversion rate into pathogenic T cells. Importantly, the decreased alloreactivity of Sirt-1-deficient T cells did not impair graft-versus-leukemia (GVL) activity in tumor models. Strikingly, transient inhibition of Sirt-1 with Ex-527 significantly prolonged the survival of recipients with no signs of tumor relapse. In agreement with aGVHD models, Sirt-1 deficiency in T cells resulted in cGVHD attenuation, which was associated with reduced Tfh generation and modulation of donor B-cell responses manifested by reduction in B-cell activation and plasma cell differentiation. Thus, Sirt-1 serves a promising therapeutic target for the prevention and treatment of GVHD.
Material and methods
Mice
C57BL/6 (B6, H-2b), BALB/c (H-2d), and B6DF1 (B6 x DBA2) F1 (H-2b/d) were purchased from the National Cancer Institute (Frederick, MD). B10.D2 (H-2d) mice were purchased from The Jackson Laboratory (Bar Harbor, ME). T-cell conditional Sirt-1−/− KO mice (Sirt-1fl/fl CD4-Cre, H-2b) on B6 background were used, in which both CD4 and CD8 T cells are deficient for Sirt-1, as CD4Cre is expressed in late double-negative to double-positive stage leading to deletion of floxed genes in both CD4 and CD8 T cells. The breeder of Sirt-1fl/fl xCD4-Cre, BALB/b, and Rag1−/− B6 mice were purchased from The Jackson Laboratory and bred in the mouse breeding facility at the Medical University of South Carolina (Charleston, SC). Animals were maintained in pathogen-free facilities in the American Association for Laboratory Animal Care–accredited Animal Resource Center at the Medical University of South Carolina. All mice procedures were approved by the Institutional Animal Care and Use Committee of the Medical University of South Carolina.
aGVHD mouse models
Recipient female mice (8-10 weeks old) were lethally irradiated at 700 cGy (BALB/c recipients) or 1100 cGy (BALB.b or BDF1 recipients), using an X-RAD 320 irradiator (Precision X-ray Inc., North Branford, CT). Irradiated mice were transplanted with 5 × 106 per mouse T-cell depleted bone marrow (TCD-BM) plus 0.7 × 106 CD25-depleted T cells for BALB/c or 2.5 × 106 CD25-depleted T cells for BALB.b or 3 × 106 CD25-depleted T cells for BDF1 model. Recipient survival, body weights, and clinical scores were monitored for 60 to 80 days. To inhibit Sirt-1 activity in vivo, Ex-527 (Selleckchem) or phosphate-buffered saline was administered at 2 mg/kg/mouse/day intraperitoneally daily from day 0 of allo-BMT for 3 weeks.
Experimental procedures
GVL models, cGVHD models, T-cell purification, and T-cell depleted bone marrow, antibodies and flow cytometry, in vitro T-cell polarization, in vitro and in vivo mixed lymphocyte reactions, in vitro and in vivo iTreg stability assay, lymphocyte isolation from recipient liver, histological analysis, DNA methylation assay, human Sirt-1 activity assay, and western blotting analysis are described in previously published work20-27 and are also detailed in the supplemental Methods, available on the Blood Web site.
Statistics
For comparison of recipient survival and tumor mortality rate in all experiments, the log-rank (Mantel-Cox) test (GraphPad Prism software, version 7) was used to determine statistical significances. To compare clinical scores, cell frequency, cytokine, and immune cell marker expressions, 2-tailed Student t test was performed to determine any statistical differences between 2 groups. For multiple pairwise comparisons between conditions, ordinary 1-way analysis of variance with Sidak multiple comparisons test was used to adjust P values. P < .05 was considered statistically significant.
Results
Sirt-1 regulates T-cell proliferation, Th1 differentiation, and aGVHD pathogenesis
Sirt-1 was previously shown to play a critical role in mediating autoimmune diseases such as EAE and colitis. However, 2 independent reports11,13 employing either germline or conditional Sirt-1 KO in T cells showed contradicting results. In the current study, we aimed to examine the role of Sirt-1 in T-cell responses to allo-antigen and how this influenced their pathogenicity during the induction of GVHD. We initially evaluated Sirt-1 activity in human T cells after being stimulated with allogeneic dendritic cells in vitro, which mimics activated donor T cells in patients with GVHD. We observed that that Sirt-1 activity was markedly increased after allogeneic stimulation (supplemental Figure 1A). Furthermore, we have tested the effect of Ex-527 on human T-cell proliferation after stimulation with allogeneic dendritic cells and found that Sirt-1 inhibition by Ex-527 significantly reduced T-cell proliferation (supplemental Figure 1B). These results provide the evidence that human T cells enhanced Sirt-1 activity in allogeneic response, and Sirt-1 activity is required for optimal T-cell activation and proliferation. To understand how Sirt-1 regulates T-cell allo-responses, we subsequently focused on preclinical studies by using conditional Sirt-1 KO mice that were generated by crossing Sirt-1flox/flox with CD4-Cre. CD4 (Cre) is expressed in late double-negative to double-positive stages that leading to deletion of floxed genes in both CD4 and CD8 T cells.28,29 Therefore, Sirt-1fl/fl CD4Cre+ mice have Sirt-1 deficiency in both CD4 and CD8 T cells.
We further investigated the effect of Sirt-1 deletion on immunological phenotype. Sirt-1 deficiency in T cells had no effect on T-cell development, as a similar frequency of double-positive T cells was observed in the thymus of wild-type (WT) and Sirt-1 knock-out mice. As expected, an elevated protein acetylation level was detected in the thymocytes of Sirt-1−/− mice, confirming previous reports documenting the deacetylation function of Sirt-1 (supplemental Figure 2A-B). The frequencies of CD4, CD8, and associated memory T-cell subsets including naive (CD44-CD62L+), central memory (CD44+CD62L+), and effector memory T cells (CD44+CD62L−), nTregs (CD4+CD25+Foxp3), and B cells were also similar between WT and Sirt-1−/− mice, as previously reported (supplemental Figure 2C-E).14 To test the role of Sirt-1 in T-cell differentiation, CD4+CD25− T cells were stimulated under different polarization conditions. We observed that Sirt1−/− CD4 T cells were less polarized into either Th1 or Th17 subsets compared with WT T cells, suggesting that Sirt-1 is required for naive T cells to optimally differentiate into Th1 and Th17 cells. However, we found that Sirt-1 is dispensable for iTreg differentiation in vitro (supplemental Figure 3A).
We further tested the effect of Sirt-1 in T-cell responses to alloantigen and found that Sirt-1−/− T cells, particularly CD4 T cells, had a reduced ability to proliferate in vitro after allogeneic stimulation, reflected by a significantly decreased carboxyfluorescein diacetate succinimidyl ester (CFSE) dilution. Moreover, Sirt-1−/− CD4, but not CD8, T cells produced dramatically less interferon (IFN)-γ on stimulation compared with WT counterparts (supplemental Figure 3B). Consistent with in vitro stimulation, transfer of Sirt1−/− T cells into lethally irradiated BALB/c mice resulted in a lower ability to proliferate and produce cytokines in vivo, as reflected by a lower proportion of CFSE-diluted cells and IFN-γ expression (Figure 1A-B).
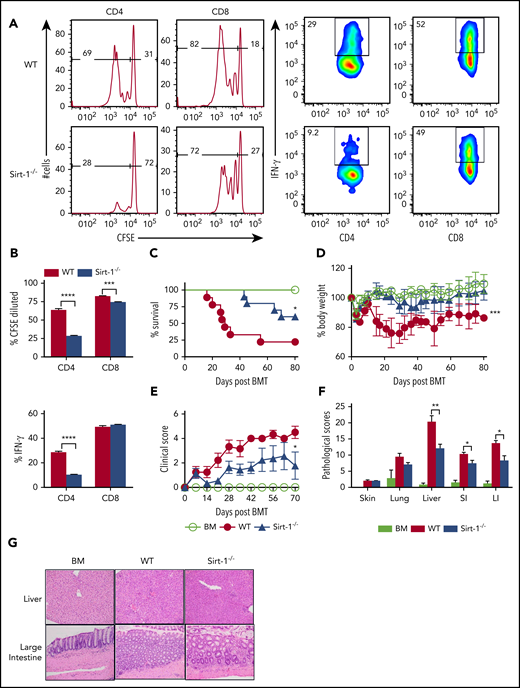
Sirt-1 regulates T-cell proliferation, differentiation, and aGVHD pathogenicity. (A) Purified T cells from either WT or Sirt-1−/− donors (H-2b) were labeled with CFSE and transferred into lethally irradiated BALB/c (H-2d) mice at 2 × 106 cells per mouse. Three days after cells transfer, recipient spleens were harvested and analyzed by flow cytometry. Representative figures and percentages of CFSE and IFN-γ are shown on gated live cells followed by H-2b+CD4+ or CD8+ cells. (B) Average percentages of CFSE-diluted and IFN-γ+ cells are shown on gated live donor CD4+ or CD8+ T cells in recipient spleen (n = 5 mice/group). (C-E) Lethally irradiated BALB/c (700 cGy) mice underwent transplantation with 5 × 106 TCD-BM per mouse plus 0.7 × 106 CD25-depleted T cells. (C) Survival, (D) bodyweight loss, and (E) clinical scores were monitored. (F-G) Tissues from BALB/c recipients were collected on day 14 after allo-BMT and analyzed for pathology. (G) Hematoxylin and eosin staining of representative pictures of livers and large intestines are shown (n = 10 mice/group). Original magnification ×200. Log-rank (Mantel-Cox) test was used to analyze the survival curve. Student t test was used for statistical analysis. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Sirt-1 regulates T-cell proliferation, differentiation, and aGVHD pathogenicity. (A) Purified T cells from either WT or Sirt-1−/− donors (H-2b) were labeled with CFSE and transferred into lethally irradiated BALB/c (H-2d) mice at 2 × 106 cells per mouse. Three days after cells transfer, recipient spleens were harvested and analyzed by flow cytometry. Representative figures and percentages of CFSE and IFN-γ are shown on gated live cells followed by H-2b+CD4+ or CD8+ cells. (B) Average percentages of CFSE-diluted and IFN-γ+ cells are shown on gated live donor CD4+ or CD8+ T cells in recipient spleen (n = 5 mice/group). (C-E) Lethally irradiated BALB/c (700 cGy) mice underwent transplantation with 5 × 106 TCD-BM per mouse plus 0.7 × 106 CD25-depleted T cells. (C) Survival, (D) bodyweight loss, and (E) clinical scores were monitored. (F-G) Tissues from BALB/c recipients were collected on day 14 after allo-BMT and analyzed for pathology. (G) Hematoxylin and eosin staining of representative pictures of livers and large intestines are shown (n = 10 mice/group). Original magnification ×200. Log-rank (Mantel-Cox) test was used to analyze the survival curve. Student t test was used for statistical analysis. *P < .05; **P < .01; ***P < .001; ****P < .0001.
To investigate the role of Sirt-1 in mediating aGVHD, we first employed a major histocompatibility complex (MHC)–mismatched (B6 to BALB/c) BMT model. CD25-depleted Sirt-1−/− T cells had a significantly reduced ability to induce aGVHD compared with WT T cells, reflected by improved survival as well as lower body weight loss and clinical scores of recipient mice after allo-BMT (Figure 1C-E). Consistently, Sirt-1−/− T cells also had a marked reduction in aGVHD severity than WT T cells in a MHC-matched (B6 to BALB.b) BMT model (supplemental Figure 4A-C). This reduction in GVHD severity was verified by pathologic analysis in recipient liver and small and large intestines (Figure 1F-G). Collectively, these data suggested that Sirt-1 contributes to T-cell activation and pathogenicity during development of aGVHD.
Sirt-1 inhibits iTreg differentiation and promotes IFN-γ production after allo-BMT
Th1 and Tc1 subsets have been implicated in mediating aGVHD pathogenesis, whereas iTregs play a protective role in disease development.30,31 We therefore sought to determine the fate of Sirt-1−/− T cells in recipients after allo-BMT. Unlike iTreg generation in vitro (supplemental Figure 3A), Sirt-1−/− CD4 and CD8 T cells in the spleen had rapidly differentiated into iTregs 14 days after BMT (Figure 2A-B). These T cells also exhibited significantly lower IFN-γ production compared with WT counterparts (Figure 2C). In contrast, Sirt-1−/− CD4 T cells secreted higher levels of IL-4, which was previously shown to inhibit Th1 responses (Figure 2D).32 The frequency of IL-17 producing CD4 T cells in the spleen was not different between these 2 groups (Figure 2D). Consistent with T-cell differentiation in spleen, we observed enhanced iTreg generation in the liver of recipients transplanted with Sirt-1−/− T cells (Figure 2E-G). We also found the liver to contain substantially decreased numbers of T cells in Sirt-1−/− recipients (Figure 2F), and those cells had markedly reduced IFN-γ and TNF-α production compared with WT T cells (Figure 2H; supplemental Figure 5A-B). Furthermore, pro-inflammatory TNF-α was significantly reduced, but anti-inflammatory IL-4 and IL-10 were significantly increased, in the serum of the recipients transplanted with Sirt-1−/− in comparison with WT T cells (Figure 2I). Collectively, these results demonstrate that Sirt-1 promotes T-cell activation and inflammatory cytokine production during allo-responses, and thus enhances T-cell pathogenicity in the development of aGVHD after allo-BMT.
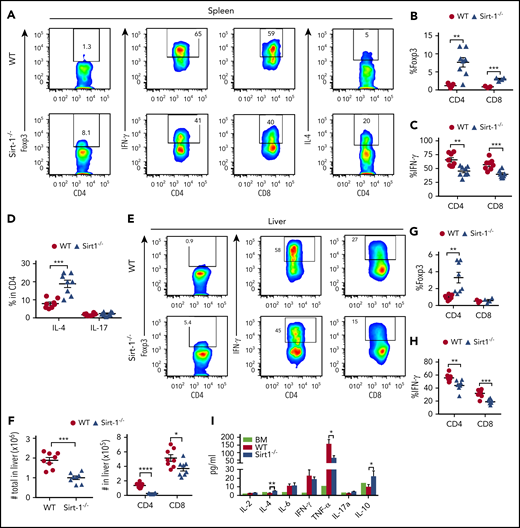
Sirt-1 inhibits iTreg differentiation after allo-BMT. Two weeks after allo-BMT (B6 to BALB/c model), spleen and liver of the recipients were harvested and analyzed. (A-D) Representative dot plots and the average percentages of Foxp3, IFN-γ, IL-4, and IL-17 expressions on gated donor CD4+ or CD8+ T cells from spleen are shown. (E-H) Representative dot plots and the frequency of Foxp3 and IFN-γ+ cells gated on donor CD4+ or CD8+ T cells from liver are shown. (F) The absolute number of total cells recovered from liver and absolute numbers of donor CD4+ and CD8+ T cells are depicted. (I) Cytokine analyses of mice serum on day 14 are shown (n = 8 mice/group). Student t test was used for statistical analysis. *P < .05; **P < .01; ***P < .001.
Sirt-1 inhibits iTreg differentiation after allo-BMT. Two weeks after allo-BMT (B6 to BALB/c model), spleen and liver of the recipients were harvested and analyzed. (A-D) Representative dot plots and the average percentages of Foxp3, IFN-γ, IL-4, and IL-17 expressions on gated donor CD4+ or CD8+ T cells from spleen are shown. (E-H) Representative dot plots and the frequency of Foxp3 and IFN-γ+ cells gated on donor CD4+ or CD8+ T cells from liver are shown. (F) The absolute number of total cells recovered from liver and absolute numbers of donor CD4+ and CD8+ T cells are depicted. (I) Cytokine analyses of mice serum on day 14 are shown (n = 8 mice/group). Student t test was used for statistical analysis. *P < .05; **P < .01; ***P < .001.
Sirt-1 enhances T-cell alloreactivity through p53 deacetylation
We next sought to inhibit Sirt-1 pharmacologically for translational purposes. The Sirt-1 inhibitor, Ex-527, was shown to have high specificity for Sirt-1 and to enhance cell cycle arrest at the G1 phase in cancer cells.33 Ex-527 was also demonstrated to promote iTregs while inhibiting Th17 differentiation.13,15 To test the effect of Ex-527 on T-cell allo-responses, we stimulated T cells with allogeneic antigen-presenting cells (APCs) in vitro, and found that Ex-527 significantly reduced CD4 T-cell proliferation and IFN-γ production to a comparable level of Sirt-1−/− T cells. Given Ex-527 had no additional effect on Sirt-1−/− T cells, we interpret that the effect of Ex-527 was specific to T cells rather than to APCs (Figure 3A-B).
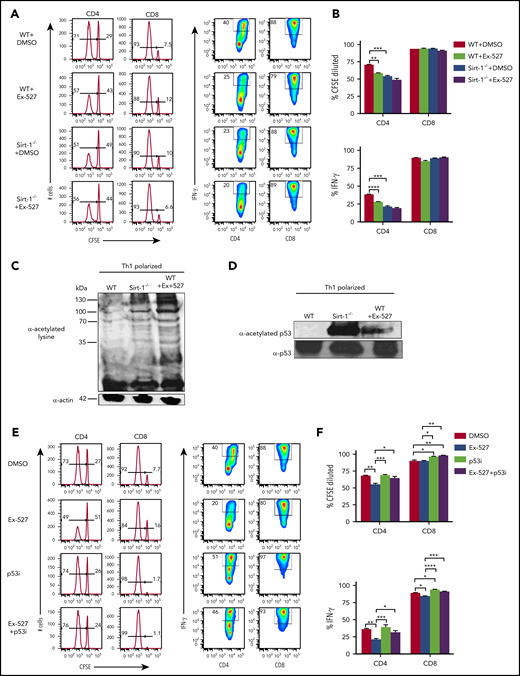
Sirt-1 enhances T-cell alloreactivity through p53 deacetylation. (A-B) Purified WT or Sirt-1−/− T cells were labeled with CFSE and cocultured with T-cell-depleted splenocytes as APCs from BALB/c mice for 5 days in the presence of either dimethyl sulfoxide or Ex-527 (10 µg/mL). Cells were restimulated with phorbol 12-myristate 13-acetate and ionomycin for cytokine measurement. Average percentages of CFSE-diluted and IFN-γ+ gated on donor CD4 or CD8 T cells are shown. (C) CD4+ T cells isolated from WT or Sirt-1−/− or WT T cells treated with Ex-527 were polarized into Th1 cells in the presence of syngeneic APCs with 1 μg/mL anti-mouse CD3ε (clone 145-2C11); 10 ng/mL mIL-12, and 1 ng/mL mIFN-γ were used for Th1 polarization. For western blot analysis, polarized cells were pretreated with 2 μM Trichostatin A (Sigma Aldrich) for 45 minutes to induce basal protein acetylation for detection of global acetylation. (D) Purified CD4 T cells from WT or Sirt-1−/− or Ex-527-treated cells were polarized under Th1 condition for 3 days and analyzed by western blot analysis for α-acetylated p53 and total p53. (E-F) T cells isolated from WT were labeled with CFSE and cocultured with allogeneic APCs for 5 days in the presence of Ex-527 or Pifithrin-μ, p53 inhibitor (10 μg/mL each) or in combinations. The frequency of CFSE diluted and IFN-γ gated on donor CD4 or CD8 T cells were analyzed. Data represent mean ± standard error of the mean. Ordinary 1-way analysis of variance with Sidak multiple comparisons test was used for statistical analysis (n = 6 per group). *P < .05; **P < .01; ***P < .001; ****P < .0001.
Sirt-1 enhances T-cell alloreactivity through p53 deacetylation. (A-B) Purified WT or Sirt-1−/− T cells were labeled with CFSE and cocultured with T-cell-depleted splenocytes as APCs from BALB/c mice for 5 days in the presence of either dimethyl sulfoxide or Ex-527 (10 µg/mL). Cells were restimulated with phorbol 12-myristate 13-acetate and ionomycin for cytokine measurement. Average percentages of CFSE-diluted and IFN-γ+ gated on donor CD4 or CD8 T cells are shown. (C) CD4+ T cells isolated from WT or Sirt-1−/− or WT T cells treated with Ex-527 were polarized into Th1 cells in the presence of syngeneic APCs with 1 μg/mL anti-mouse CD3ε (clone 145-2C11); 10 ng/mL mIL-12, and 1 ng/mL mIFN-γ were used for Th1 polarization. For western blot analysis, polarized cells were pretreated with 2 μM Trichostatin A (Sigma Aldrich) for 45 minutes to induce basal protein acetylation for detection of global acetylation. (D) Purified CD4 T cells from WT or Sirt-1−/− or Ex-527-treated cells were polarized under Th1 condition for 3 days and analyzed by western blot analysis for α-acetylated p53 and total p53. (E-F) T cells isolated from WT were labeled with CFSE and cocultured with allogeneic APCs for 5 days in the presence of Ex-527 or Pifithrin-μ, p53 inhibitor (10 μg/mL each) or in combinations. The frequency of CFSE diluted and IFN-γ gated on donor CD4 or CD8 T cells were analyzed. Data represent mean ± standard error of the mean. Ordinary 1-way analysis of variance with Sidak multiple comparisons test was used for statistical analysis (n = 6 per group). *P < .05; **P < .01; ***P < .001; ****P < .0001.
To investigate the molecular mechanisms by which Sirt-1 regulates T-cell responses, we initially compared total protein acetylation of T cells. We found that Sirt-1−/− Th1 cells remained hyperacetylated compared with WT Th1 cells. Consistently, Sirt-1 inhibition with Ex-527 also enhanced total acetylation of WT Th1 cells (Figure 3C) Furthermore, similar results were observed in T cells isolated from the recipient mice 14 days after allo-BMT (supplemental Figure 5C). As p53 protein was previously identified as a Sirt-1 target in cancer cells,34-37 we further evaluated the acetylation level of p53 in Sirt1−/− CD4 T cells under Th1 polarizing conditions. In agreement with the previous reports, we also found that Sirt-1 deficiency or Sirt-1 inhibition substantially increased acetylation of p53 in Sirt1−/− and Ex-527-treated Th1 cells (Figure 3D). Because hyperacetylation of p53 is associated with stability, and thereby functions to inhibit T-cell proliferation and activation,38-40 we hypothesized that Sirt-1 regulates T-cell alloreactivity through p53 signaling. To test that hypothesis, we used a p53 inhibitor, Pifithrin-μ, a specific inhibitor known to reduce the affinity of p53 binding to Bcl-xL and Bcl-2, antiapoptotic molecules.41,42 Indeed, inhibition of p53 essentially abolished the effect of EX-527 on T-cell proliferation and IFN-γ production (Figure 3E-F). These data indicated that Sirt-1 regulates T-cell alloreactivity through deacetylation of p53 signaling.
Sirt-1 deacetylates Foxp3 protein and reduces iTreg stability
As Foxp3+ Tregs were derived from Sirt-1−/− T cells in allogeneic recipients (Figure 2A-B), and Sirt-1 did not affect the generation of iTregs from naive T cells (supplemental Figure 3A), we hypothesized that the absence of Sirt-1 may enhance Foxp3 stability in iTregs, and thus bolster their suppressive function and subsequently alleviate GVHD.15,43,44 We addressed this by performing in vitro and in vivo iTreg stability assays. Given that the most dramatic effect was seen in CD4 iTreg generation after allo-BMT, we generated CD4 iTregs in vitro from Sirt-1−/− CD4 T cells and then enriched CD25high cells for our assays (Figure 4A). We first confirmed that acetylation of Sirt-1−/− CD4 iTregs was elevated compared with WT CD4 iTregs (Figure 4B). Because STAT5 signaling was previously shown to be increased in Tregs compared with conventional T cells, and was associated with Treg stability,45,46 we evaluated phospho-STAT5 (pSTAT5) levels and found that they were significantly elevated in Sirt-1−/− CD4 iTregs or WT CD4 iTregs incubated with EX-527 (Figure 4C). Furthermore, Sirt-1−/− CD4 iTregs maintained higher Foxp3 expression compared with WT CD4 iTregs in either conditions favoring Tregs (with IL-2 alone) or Th1-favoring conditions (IL-2 plus IL-12; Figure 4D).
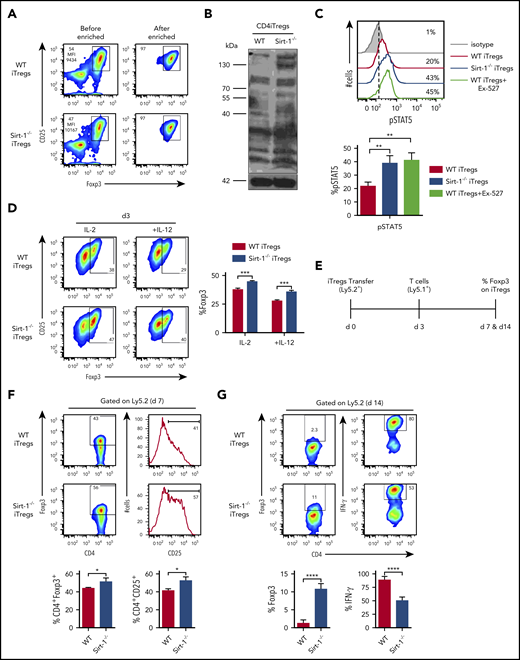
Sirt-1 deacetylates Foxp3 and decreases iTregs stability. (A) Allogeneic CD4 iTregs were generated in vitro by coculturing naive CD4 T cells with allogeneic dendritic cells in the presence of IL-2, TGF-β, and retinoic acid for 5 days. CD4 iTregs were enriched for CD25hi cells. (B) Enriched CD25+ CD4iTregs were analyzed by western blot analysis for detection of global acetylation. (C) Five-day in vitro-generated CD4 iTregs were restimulated with IL-2 and analyzed for pSTAT5 expression. (D) Enriched CD4 iTregs were cocultured with allogeneic APCs in the presence of IL-2 or IL-2+IL-12 for 3 days. Percentage Foxp3 retention was analyzed on day 3 (n = 4). (E) Experimental scheme: lethally irradiated BALB/c mice were adoptively transferred with 5 × 106 Rag1−/− BM and 0.5-1 × 106 CD4 iTregs (Ly5.2). On day 3, 0.5-2 × 106 CD25-depleted T-cells from B6Ly5.1 congenic mice were injected to induce GVHD in recipients. On day 7 or 14 after allo-BMT, spleen was harvested and analyzed. (F) Percentages of Foxp3 and CD25 expressions on CD4iTregs analyzed on day 7 (gated on Ly5.2+CD4+) are shown. (G) Percentages of Foxp3 and IFN-γ expressions of transferred CD4 iTregs are shown (n = 4-5 mice per group). Data represent the mean ± standard error of the mean. Student t-test was used for statistical analysis. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Sirt-1 deacetylates Foxp3 and decreases iTregs stability. (A) Allogeneic CD4 iTregs were generated in vitro by coculturing naive CD4 T cells with allogeneic dendritic cells in the presence of IL-2, TGF-β, and retinoic acid for 5 days. CD4 iTregs were enriched for CD25hi cells. (B) Enriched CD25+ CD4iTregs were analyzed by western blot analysis for detection of global acetylation. (C) Five-day in vitro-generated CD4 iTregs were restimulated with IL-2 and analyzed for pSTAT5 expression. (D) Enriched CD4 iTregs were cocultured with allogeneic APCs in the presence of IL-2 or IL-2+IL-12 for 3 days. Percentage Foxp3 retention was analyzed on day 3 (n = 4). (E) Experimental scheme: lethally irradiated BALB/c mice were adoptively transferred with 5 × 106 Rag1−/− BM and 0.5-1 × 106 CD4 iTregs (Ly5.2). On day 3, 0.5-2 × 106 CD25-depleted T-cells from B6Ly5.1 congenic mice were injected to induce GVHD in recipients. On day 7 or 14 after allo-BMT, spleen was harvested and analyzed. (F) Percentages of Foxp3 and CD25 expressions on CD4iTregs analyzed on day 7 (gated on Ly5.2+CD4+) are shown. (G) Percentages of Foxp3 and IFN-γ expressions of transferred CD4 iTregs are shown (n = 4-5 mice per group). Data represent the mean ± standard error of the mean. Student t-test was used for statistical analysis. *P < .05; **P < .01; ***P < .001; ****P < .0001.
To investigate the role of Sirt-1 in Foxp3 stability in vivo, we transferred iTregs into allogeneic recipients during the early stages of GVHD development (Figure 4E). Four days after T effector cell (Ly5.1+) infusion into lethally irradiated recipients, similar frequencies of WT and Sirt-1−/− CD4 iTregs (Ly5.2+) were found in recipient spleens. However, we observed that Sirt-1−/− iTregs maintained a higher level of Foxp3 and IL-2Rα expressions than WT CD4 iTregs, suggesting that the absence of Sirt-1 increased Foxp3 stability and reduced the conversion iTregs into IFN-γ-producing Th1 cells (Figure 4F).47 By day 14, we observed that WT iTregs essentially lost their Foxp3 expression, but 10% to 15% of Foxp3 expression was retained in Sirt-1−/− iTregs. Moreover, transferred Sirt-1−/− iTregs had dramatically less conversion into IFN-γ+ cells compared with WT counterparts (Figure 4G). Given the methylation status of Foxp3 impacts, we further measured Foxp3 methylation of Sirt-1−/− CD4 iTregs and found that iTregs generated from WT or Sirt-1−/− T cells had similar levels of Foxp3 demethylation (supplemental Figure 6A-B). These results are consistent with a previous report48 that loss of Sirt-1 did not alter Treg-specific demethylated region methylation status in Tregs. Instead, this study pointed out that Sirt-1 functions in posttranslational modification, where Sirt-1 hyperacetylated Foxp3 protein and lead to an increased iTreg-stability. Acetylated Foxp3 molecules have previously been shown to be more stable than those with hypo-acetylation, as this modification prevents ubiquitylation at targeted lysine residues, and thus become resistant to proteasomal degradation.49
Pharmacological inhibition of Sirt-1 alleviates aGVHD while preserving GVL activity
To investigate the potential for clinical translation, we next evaluated the efficacy of Ex-527 in aGVHD prevention, using an MHC-mismatched model (B6 to BALB/c). Treatment of recipients with Ex-527 resulted in improved clinical scores and significantly prolonged survival compared with vehicle control (Figure 5A-B). To evaluate the effect of Sirt-1 inhibition on the GVL activity, we employed 2 tumor models, B6 to BALB/c with MLL-AF9-GFP tumor and a haploidentical B6 to BDF1 model with P815 tumor. BALB/c recipients transplanted with WT T cells and treated with vehicle developed severe GVHD and died early after BMT with no detectable signs of tumor relapse in the blood on days 20 and 30. In contrast, the recipients transplanted with Sirt-1−/− T cells displayed alleviated GVHD with significantly prolonged survival. Nevertheless, some of the recipients had indications of tumor relapse with an increased percentage of GFP+ cells in the blood. Strikingly, the recipients transplanted with WT T cells and treated with Ex-527 had significantly improved survival with no signs of tumor relapse, indicating that Sirt-1 inhibition could attenuate GVHD while preserving GVL activity (Figure 5C-D). Similar results were observed when nTregs were included in the donor grafts (supplemental Figure 7A-C).
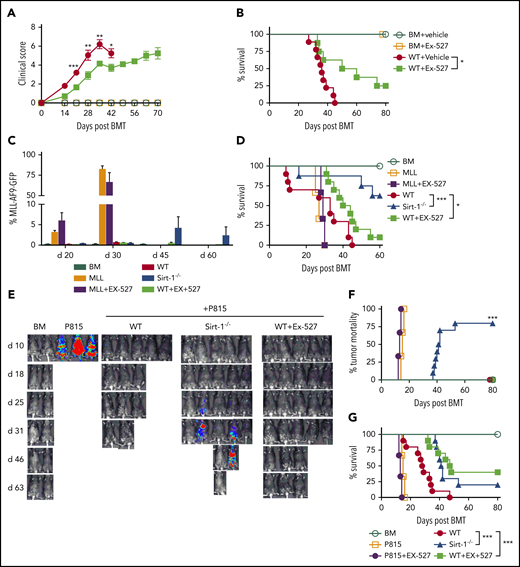
Pharmacological inhibition of Sirt-1 with Ex-527 alleviates aGVHD while preserving the GVL activity. (A-B) GVHD experiments: Lethally irradiated (700 cGy) BALB/c mice underwent transplantation with 5 × 106 TCD-BM alone or plus 0.7 × 106 CD25− T cells per mouse isolated from WT and administered with either phosphate-buffered saline or 2 mg/kg Ex-527/mouse/day daily for 3 weeks. (A) GVHD clinical score. (B) Survival (n = 10 mice/group). (C-G) GVL experiments: (C) Bar graph shows quantified percentages of MLL-AF9-GFP in the recipients’ peripheral blood on days 20, 30, 45, and 60 (n = 10 mice/group). (D) Survival of recipient mice was monitored. (E-G) Lethally irradiated (1100 cGy, split dose) BDF1 recipients were transplanted with 5 × 106 TCD-BM and 5 × 103 P815 mastocytoma plus 3 × 106 CD25− T cells. Recipients were monitored for tumor burden. (E) Representative images of tumor growth measured by bioluminescent imaging. (F) Tumor mortality and (G) mouse survival were monitored. Data were combined from 2 independent experiments (n = 10 mice/group). Log-rank (Mantel-Cox) test was used to compare the tumor mortality and survival. *P < .05; **P < .01; ***P < .001.
Pharmacological inhibition of Sirt-1 with Ex-527 alleviates aGVHD while preserving the GVL activity. (A-B) GVHD experiments: Lethally irradiated (700 cGy) BALB/c mice underwent transplantation with 5 × 106 TCD-BM alone or plus 0.7 × 106 CD25− T cells per mouse isolated from WT and administered with either phosphate-buffered saline or 2 mg/kg Ex-527/mouse/day daily for 3 weeks. (A) GVHD clinical score. (B) Survival (n = 10 mice/group). (C-G) GVL experiments: (C) Bar graph shows quantified percentages of MLL-AF9-GFP in the recipients’ peripheral blood on days 20, 30, 45, and 60 (n = 10 mice/group). (D) Survival of recipient mice was monitored. (E-G) Lethally irradiated (1100 cGy, split dose) BDF1 recipients were transplanted with 5 × 106 TCD-BM and 5 × 103 P815 mastocytoma plus 3 × 106 CD25− T cells. Recipients were monitored for tumor burden. (E) Representative images of tumor growth measured by bioluminescent imaging. (F) Tumor mortality and (G) mouse survival were monitored. Data were combined from 2 independent experiments (n = 10 mice/group). Log-rank (Mantel-Cox) test was used to compare the tumor mortality and survival. *P < .05; **P < .01; ***P < .001.
Consistently, in BDF1 recipients with P815 tumors, the majority of the recipients transplanted with WT T cells developed severe GVHD and died within 40 days. Similar to observations in the MHC-mismatched model, some of the recipients who received Sirt-1−/− grafts had tumor relapse detected on day 30 after allo-BMT. Importantly, Sirt-1 inhibition with Ex-527 significantly prolonged the survival of recipients with no signs of tumor growth, which further indicated that pharmacological blockade of Sirt-1 in T cells with Ex-527 did not impair GVL activity (Figure 5E-G). Ex-527 had no direct effects on tumor growth, as recipients of BM alone with tumors and treated with the inhibitor succumbed to tumor relapse in a manner similar to that of those with vehicle control. Taken together, pharmacological blockade of Sirt-1 with Ex-527 alleviated GVHD without impairing T-cell-mediated GVL activity.
Sirt-1 expression in T cells modulates B-cell responses during cGVHD development
TCR and BCR signaling play central roles in mediating cGVHD. B-cell activation and Tfh and plasma cell differentiation contribute to cGVHD development.50,51 We hypothesized that Sirt-1-deficient T cells would reduce T- and B-cell interactions, hence resulting in less B-cell activation and pathogenic responses. To test our hypothesis, we used a B6 to BALB/c model and transplanted whole splenocytes containing WT or Sirt-1−/− T cells into recipients. Consistent with aGVHD outcomes, the recipients of Sirt-1 −/− splenocytes had significantly reduced body weight loss and clinical scores (Figure 6A-B). Pathology scores were markedly improved in GVHD target organs of the recipients with Sirt-1−/− grafts, such as in the skin, lung, liver, and small and large intestines (Figure 6C-D). Furthermore, we have measured fibrosis and observed higher collagen deposition in the skins and lungs of the recipients with WT grafts compared with those of BM alone or Sirt-1−/− graft on day 60 post allo-BMT. These results confirm that cGVHD severity was alleviated in the recipients of Sirt-1-deficient T cells compared with those of WT counterparts (supplemental Figure 8).
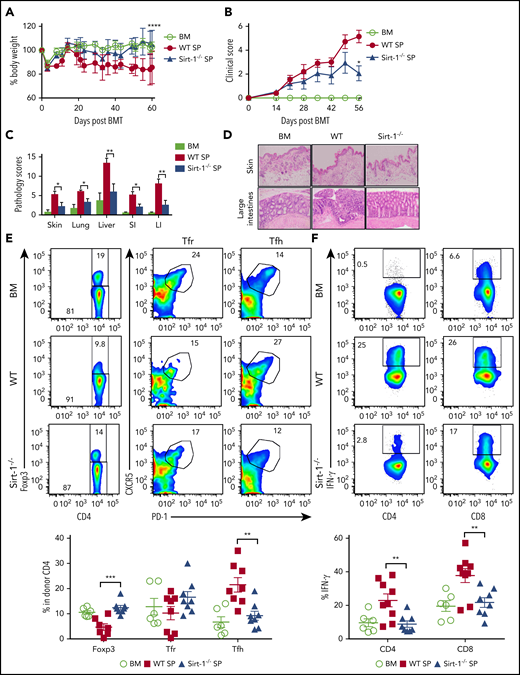
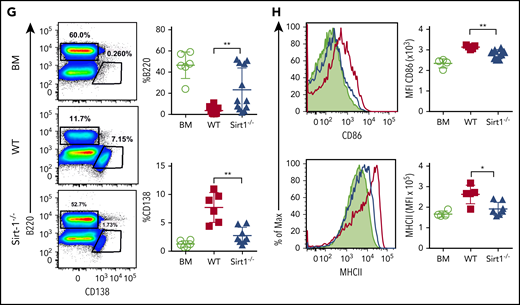
Sirt-1 modulates T- and B-cell activation in cGVHD. Lethally irradiated (700 cGy) BALB/c mice underwent transplantation with 5 × 106 TCD-BM alone or plus 5 × 105 CD25− splenocytes per mouse isolated from WT or Sirt-1−/− donors. (A) Body weight loss. (B) cGVHD clinical and (C) pathology scores. (D) Representative pictures of skin and large intestine biopsies stained with hematoxylin and eosin (original magnification ×200). (E) Foxp3 expression on gated donor CD4 in spleen and frequency of follicular Treg cells (Foxp3+CXCR5+PD-1+) and Tfh (Foxp3−CXCR5+PD-1+) were determined on day 60. (F) IFN-γ expression was shown on gated donor CD4 and CD8 T cells. (G) Splenic B cells were analyzed for B220+, B220−CD138+, and (H) activation markers, CD86 and MHCII. Data were combined from 2 independent experiments (n = 6-10 mice/group). Log-rank (Mantel-Cox) test was used to analyze body weight, and Student t test was used for statistical analysis. *P < .05; **P < .01; ***P < .001; ****P < .0001.
Sirt-1 modulates T- and B-cell activation in cGVHD. Lethally irradiated (700 cGy) BALB/c mice underwent transplantation with 5 × 106 TCD-BM alone or plus 5 × 105 CD25− splenocytes per mouse isolated from WT or Sirt-1−/− donors. (A) Body weight loss. (B) cGVHD clinical and (C) pathology scores. (D) Representative pictures of skin and large intestine biopsies stained with hematoxylin and eosin (original magnification ×200). (E) Foxp3 expression on gated donor CD4 in spleen and frequency of follicular Treg cells (Foxp3+CXCR5+PD-1+) and Tfh (Foxp3−CXCR5+PD-1+) were determined on day 60. (F) IFN-γ expression was shown on gated donor CD4 and CD8 T cells. (G) Splenic B cells were analyzed for B220+, B220−CD138+, and (H) activation markers, CD86 and MHCII. Data were combined from 2 independent experiments (n = 6-10 mice/group). Log-rank (Mantel-Cox) test was used to analyze body weight, and Student t test was used for statistical analysis. *P < .05; **P < .01; ***P < .001; ****P < .0001.
In mechanistic studies, Sirt-1−/−T cells displayed a significant increase in iTreg generation with lower frequencies of splenic Tfh cells compared with WT counterparts (Figure 6E). Follicular Treg cells were previously shown to produce IL-10 and can inhibit Tfh generation.18,19 The recipients of Sirt-1−/− grafts had reduced frequencies of IFN-γ-producing T cells, indicating less T-cell activation during cGVHD onset (Figure 6F). We further investigated how Sirt-1 deficiency in T cells affects B-cell responses during cGVHD. Splenic B-cell reconstitution was increased in recipients transplanted with Sirt-1−/− grafts. These B cells were less differentiated into plasma cells, reflected by the lower percentages of B220lowCD138+ cells (Figure 6G). The reduction in cGVHD and reduced T-cell responses correlated with impaired B-cell activation, including downregulation of CD86 and MHC class II expression on splenic B cells in the recipients transplanted with Sirt-1−/− grafts (Figure 6H). These data suggest that Sirt-1 is essential for T cells to induce B-cell responses.
Treatment with Ex-527 prevents cGVHD and also attenuates established cGVHD
We next tested whether Sirt-1 blockade could ameliorate cGVHD using both MHC-mismatched B6 to BALB/c and MHC-matched B10D2 to BALB/c models. In the MHC-matched scleroderma cGVHD model (B10D2 to BALB/c), administration of Ex-527 starting at day 0 significantly reduced the severity of cGVHD reflected by clinical scores. Moreover, administration of Ex-527 from day 28 also reduced the severity of established cGVHD, albeit less profoundly (Figure 7A-B). Early treatment of Ex-527 markedly inhibited the production of IFN-γ and IL-17 by donor T cells compared with vehicle and the late treatment group (Figure 7C-D). Tfh cell subsets have been shown to express a high level of BCL6 and IL-21 cytokines, which contribute to cGVHD development. CD4 T cells from recipients treated with Ex-527 exhibited significantly lower BCL-6 expression and IL-21 production, suggesting less pathogenic CD4 T cells generated on Sirt-1 inhibition (Figure 7E-F). Consistently, the frequencies of splenic Tfh and PD-1 expression on donor CD8 T cells were also reduced in the recipients treated with Ex-527 from day 0 compared with vehicle control (Figure 7G). In agreement with genetic KO of Sirt-1 splenocytes, the inhibitor significantly improved B-cell reconstitution in the recipients treated either at early or late treatment stages. Moreover, these reconstituted donor B cells were less activated and differentiated with lower expression of CD138 and CD86 on B cells after treatment (Figure 7H). The suppressive effect of Ex-527 on cGVHD development was mimicked in the MHC-mismatched B6 to BALB/c BMT model (supplemental Figure 9). Taken together, Sirt-1 inhibition significantly reduced T- and B-cell interactions and decreased B-cell alloreactivity during cGVHD.
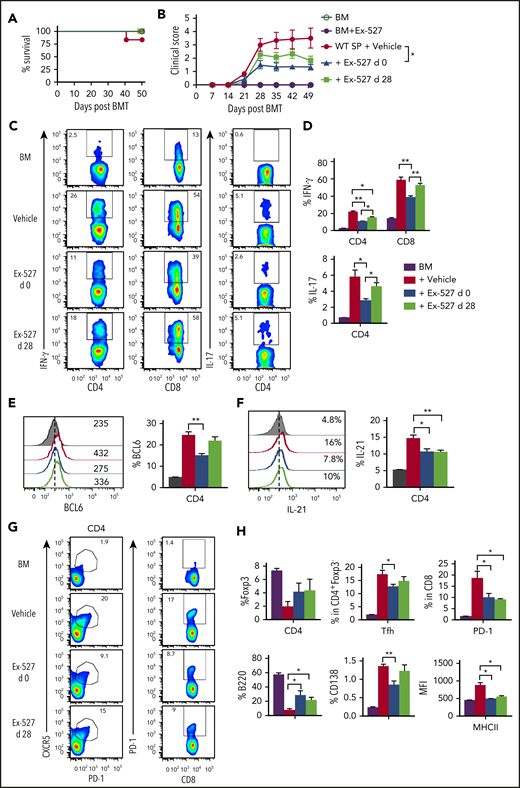
Treatment with Ex-527 attenuates cGVHD. Lethally irradiated (700 cGy) BALB/c mice underwent transplantation with 5 × 106 TCD-BM of B10.D2 BM plus 5 × 106 whole splenocytes containing CD25 per mouse isolated from WT donor. The recipients were treated either prophylactically on day 0 or day 28 posttransplant for 21 days with 2 mg/kg/mouse/day Ex-527. (A) Survival (B) cGVHD clinical scores. (C-D) Analysis of IFN-γ and IL-17 expressions on donor CD4 in spleen. (E-F) BCL6 expression and IL-21 secretion by donor CD4 were determined on day 50. (G-H) Analysis of Foxp3 expression and frequency of Tfh (Foxp3−CXCR5+PD-1+) and PD-1+CD8+ were measured on donor T cells. Splenic B cells were analyzed for B220+, B220-CD138+, and MHCII. Data were combined from 2 independent experiments (n = 5-7 mice/group). Student t test was used for statistical analysis. *P < .05; **P < .01.
Treatment with Ex-527 attenuates cGVHD. Lethally irradiated (700 cGy) BALB/c mice underwent transplantation with 5 × 106 TCD-BM of B10.D2 BM plus 5 × 106 whole splenocytes containing CD25 per mouse isolated from WT donor. The recipients were treated either prophylactically on day 0 or day 28 posttransplant for 21 days with 2 mg/kg/mouse/day Ex-527. (A) Survival (B) cGVHD clinical scores. (C-D) Analysis of IFN-γ and IL-17 expressions on donor CD4 in spleen. (E-F) BCL6 expression and IL-21 secretion by donor CD4 were determined on day 50. (G-H) Analysis of Foxp3 expression and frequency of Tfh (Foxp3−CXCR5+PD-1+) and PD-1+CD8+ were measured on donor T cells. Splenic B cells were analyzed for B220+, B220-CD138+, and MHCII. Data were combined from 2 independent experiments (n = 5-7 mice/group). Student t test was used for statistical analysis. *P < .05; **P < .01.
Discussion
Through use of strategies that involved mouse models in conjunction with a conditional genetic deletion in T cells, as well as application of a potentially translational pharmacological inhibitor, Ex-527, we demonstrated that targeting Sirt-1 in T cells significantly reduced T-cell proliferation and Th1 and Th17 differentiation, and in turn attenuated aGVHD. Mechanistic studies revealed that the loss of Sirt-1 in T cells enhanced p53 acetylation and subsequently diminished cell proliferation after allogeneic stimulation. Sirt-1 deficiency promoted iTreg differentiation and Foxp3 stability via increased its acetylation. Importantly, inhibition of Sirt-1 with Ex-527 alleviated aGVHD while preserving GVL activity. In parallel with aGVHD results, Sirt-1−/− donor T cells also had a decreased ability to induce cGVHD, which involved augmenting iTreg generation, and thereby inhibiting Tfh development. Sirt-1−/− T cells also negatively regulated B-cell activation and plasma cell differentiation, indicating that Sirt-1 is required for T cells to induce B-cell responses. Strikingly, administration of Ex-527, the Sirt-1 inhibitor, was able to potently prevent as well as treat cGVHD through reducing proinflammatory cytokine productions, such as IFN-γ, IL-17, and IL-21, as well as dampen Tfh responses, all of which have been implicated in cGVHD pathogenesis to contribute to cGVHD severity.
Zhang and coworkers11 reported that Sirt-1 is required for maintaining T-cell tolerance in EAE models, using global Sirt-1 KO mice. Sirt-1 null mice were observed to develop autoimmune disease phenotypes in addition to a shortened lifespan.11 In contrast, a recent report using a conditional KO of Sirt-1 in T cells demonstrated that Sirt-1 promoted Th17 differentiation and exacerbated EAE severity.13 Such a discrepancy could be a result of the global deletion vs targeted deletion in T cells, as total depletion of Sirt-1 may result in developmental dysregulation of multiple immune cell types. The decreased inflammatory responses associated with specific targeting of Sirt-1 in T cells was corroborated in cardiac allograft and colitis models, in which Sirt-1−/− prolonged survival in mouse models by augmenting iTreg function via Foxp3 acetylation.14,15 We observed similar results with respect to increased Foxp3 expression and enhanced iTreg stability, using conditional KO mice. However, we observed little effect on Th17 differentiation, although Th1 differentiation is known to be dominant in aGVHD. As such, we found that Sirt-1 increased IFN-γ production and enhanced T-cell proliferation likely through p53 deacetylation. Our findings support previous reports that Sirt-1 promotes inflammatory responses in autoimmune disease settings.13-15
Administration of a Sirt-1 inhibitor (Ex-527) on day 0 to BMT recipients for 3 weeks significantly prolonged mouse survival and prevented tumor relapse (Figure 5). Pharmacological inhibition of Sirt-1 not only reduced alloreactivity of donor T cells but also preserved the GVL effect. Tumor relapse was observed in recipients transplanted with Sirt-1−/− T cells, which could result from a sustained increased level of iTreg generation as a result of the loss of Sirt-1 leading to an impaired antitumor activity of donor effector T cells. Given that Ex-527 had little effect on Sirt-1−/− T cells in response to allo-stimulation in vitro (Figure 3), we reason that the inhibitor had a dominant and direct effect on donor T cells, rather than on APCs. However, we cannot exclude the effect of Sirt-1 on donor B cells, particularly in cGVHD models, as previous studies on B-cell leukemia demonstrated that Sirt-1 inhibition induced tumor cell apoptosis.52,53 Thus, the role of Sirt-1 in primary B cells during cGVHD development requires further investigation.
Sirt-1 has previously been reported to be overexpressed and enhance metastasis in several cancer cell types.54-56 Targeting Sirt-1 was shown to inhibit cancer cell growth and induce cell apoptosis. The current study reveals how Sirt-1 regulates primary T-cell responses to allo-antigen and T-cell pathogenicity in GVHD induction, likely through modulation of p53. Our data also clearly demonstrate that loss of Sirt-1 stabilizes Foxp3 consistent with that previously reported by others,57 and thus prevented Treg conversion to pathogenic T cells under inflammatory conditions, as observed in the context of GVHD. Furthermore, Sirt-1 deficiency in T cells restrained B-cell activation and maturation into plasma B cells. Collectively, inhibition of Sirt-1 diminished T-cell alloreactivity, and hence impeded GVHD pathogenesis.
From a clinical perspective, the need to treat established cGVHD is of paramount importance. Thus, we consider it of significance that Ex-527 was able to substantially alleviate ongoing cGVHD (Figure 7B). Treatment with Ex-527 on day 28 reduced IL-21 production by donor T cells, as well as improved donor B-cell reconstitution (Figure 7F-H). The therapeutic schema and dose of the inhibitor may require further optimization to achieve better outcomes when using this cGVHD treatment protocol. In conclusion, we provide concrete evidence that Sirt-1 positively regulates T-cell alloreactivity in GVHD, and further that Sirt-1 can serve as a potential therapeutic target for the control of aGVHD and cGVHD pathogenesis.
Presented in abstract form at the 59th annual meeting of the American Society of Hematology, Atlanta, GA, 9 December 2017.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Yong Jiang for his technical assistance of western blot analysis. The authors also are grateful for the technical support provided by the Department of Laboratory Animal Research, Flow Cytometry Core and Imaging Core in Hollings Cancer Center at Medical University of South Carolina. The authors thank Sophie Paczesny, Indiana University School of Medicine, for generously providing the MLL-AF9 GFP leukemic cells. This work is partially supported by grants from the National Institutes of Health, National Cancer Institute (R01s CA118116 and CA169116), National Institute of Allergy and Infectious Diseases (AI118305), and National Heart, Lung, and Blood Institute (HL137373), and SmartState Endowment in Cancer Stem Cell Biology & Therapy Program (X.-Z.Y.).
Authorship
Contribution: A.D. participated in research design and execution of experiments, analyzed and interpreted data, and wrote the manuscript; S.I. performed iTreg stability, DNA demethylation assays, and western blot analysis; P.C. performed human Sirt-1 activity assay; H.D.N. performed cytokine bead array; D.B. participated in conducting experiments and revising the manuscript; C.L. performed the pathological analysis; S.M. provided the Sirt-1 KO mice and edited and revised the manuscript; and X-Z.Y. designed research, interpreted data, and edited and revised the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Xue-Zhong Yu, Department of Microbiology and Immunology, HCC350, MSC 955, Medical University of South Carolina, 86 Jonathan Lucas St, Charleston, SC 29425-5090; e-mail: yux@musc.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal