

Key Points
CCNA1-directed T cells expanded from healthy donors show specific antileukemic activity towards CCNA1-expressing targets.
Detection of endogenous CCNA1-specific T cells in peripheral blood is associated with clinical remission in AML patients after allo-HSCT.

Abstract
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the only curative option for relapsed or refractory acute myeloid leukemia (AML). However, more than half ultimately experience disease relapse that is associated with a dismal median survival of just 6 months, highlighting the need for novel therapies. In the current study we explore the therapeutic potential of targeting cyclin A1 (CCNA1), a cancer-testis antigen that is overexpressed in malignant blasts and leukemic stem cells. We demonstrate the immunogenicity of this antigen to native T cells, with >90% of donors screened mounting a specific response. The expanded cells were Th1 polarized, polyfunctional, and cytotoxic toward CCNA1+/HLA-matched tumor cell lines. Furthermore, these cells were exquisitely specific for CCNA1 and exhibited no reactivity against other cyclin family members, including CCNA2, which shares 56% homology with CCNA1 and is ubiquitously expressed in dividing cells. Lastly, the detection of CCNA1-specific T cells in AML patients post-HSCT was associated with prolonged disease remission, suggesting the protective potential of such endogenous cells. Taken together, our findings demonstrate the feasibility of targeting CCNA1 and the potential for therapeutic benefit associated with the adoptive transfer of reactive cells.
Introduction
Despite some advances in the treatment of acute myeloid leukemia (AML), the prognosis remains poor, with a dismal 5-year overall survival (OS) of just 28.3% in the United States.1 Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the best curative option for those with high risk or relapsed disease, but treatment-related mortality and disease relapse remain problematic.2,3 Indeed, up to 50% of allo-HSCT recipients will eventually relapse, and these patients have a 1-year OS of <20%, highlighting the need for novel therapies.4,5
Adoptive T-cell transfer holds promise as a targeted approach to treat refractory hematologic malignancies. For example, CD19-directed chimeric antigen receptor (CAR)–modified T cells have produced initial response rates of up to 90% in patients with B-cell acute lymphoblastic leukemia.6 AML has also been targeted immunotherapeutically.7 Indeed, the infusion of unselected donor lymphocytes to mediate “graft-versus-leukemia” effects in the setting of AML relapse after allo-HSCT has demonstrated the disease’s susceptibility to cellular immunotherapy.8 However, response rates are low (15% to 42%), compounded by the risk of graft-versus-host disease mediated by transferred alloreactive T cells, which has led a number of groups to explore more targeted T-cell approaches.9,10 For example, potential CAR targets, including CD33 and CD123 are being evaluated, though clinical translation has proven challenging given that these molecules are expressed on normal myeloid progenitor cells, whose ablation is intolerable.11 AML cells do, however, overexpress antigens such as WT1 and PRAME, known as leukemia-associated antigens (LAAs),12 which can be targeted by the native T-cell receptor (TCR), and early-phase clinical trials have now demonstrated the safety of such LAA-directed T cells, with documented long-term remissions in patients with relapsed or high-risk AML.13
Given the clinical promise of such native TCR-targeted approaches, we sought to extend the spectrum of antigens that could be immunotherapeutically targeted by comprehensively characterizing the immunogenicity of cyclin A1 (CCNA1), a cancer-testis antigen (CTA) that is aberrantly expressed in AML. CCNA1 possesses multiple characteristics of an “ideal” T-cell target. Firstly, it is overexpressed in up to 82% of AML cells, including leukemic stem cells, thereby maximizing the potential of selectively eliminating both the tumor bulk as well as a compartment of tumor cells implicated in relapse.14,15 Clinically, CCNA1 overexpression is associated with poorer disease-free survival and thus applicable to a cohort of patients with limited therapeutic options.16 Finally, CCNA1 has been shown to be immunogenic to T cells in the context of HLA-A*0201.15
We now demonstrate the immunogenicity of CCNA1 in individuals of diverse HLA backgrounds and establish the selectivity of specific cells for malignant CCNA1-expressing targets absent cross-reactivity against normal cells expressing other cyclin family members. Finally, we profile reactive T cells in healthy donors and AML patients and demonstrate a correlation between detection and clinical outcomes in the latter, further validating CCNA1 as a relevant immunotherapeutic target for future clinical testing.
Methods
Healthy donor and patient samples
Peripheral blood mononuclear cells (PBMCs) were obtained from healthy volunteers or AML patients after allo-HSCT with informed consent on Baylor College of Medicine institutional review board–approved protocols (H-36346 and H-15280). PBMCs were used to generate dendritic cells (DCs), tumor-specific T cells, and phytohemagglutinin (PHA)–stimulated T cells (PHA blasts). PHA blasts were generated from PBMCs (0.5 × 106/mL) using PHA (5 µg/mL) (Sigma-Aldrich, St. Louis, MO) and maintained in T-cell medium (RPMI-1640, Clicks medium [Irvine Scientific, Santa Ana, CA], 5% Human AB Serum [Valley Biomedical, Winchester, VA], and GlutaMAX [Gibco, Life Technologies, Gaithersburg, MD]) supplemented with interleukin-2 (IL-2) (100 U/mL) (R&D Systems, Minneapolis, MN), which was replenished every 2 or 3 days. Cancer cell lines U266, KG1a, THP-1, and U937 were obtained from ATCC, KO52 from JCRB Cell Bank, and SET2 from DSMZ. These were grown according to manufacturers’ instructions. All cell lines were tested and identity confirmed by DNA short tandem repeat profiling (University of Arizona, Tuscon, AZ).
CCNA1-specific T-cell generation
Peptides/pepmix
For T-cell stimulation, we used a pepmix (overlapping peptide library of 102 15mers overlapping by 11 amino acids [aa]) spanning CCNA1 isoform 3 sequence, which was purchased from Genemed Synthesis (San Antonio, TX). For immunogenic peptide mapping, individual peptides were pooled into 21 minipools, each containing 3 to 11 15mer peptides, and organized such that each peptide was present in 2 minipools. Minimal epitopes were mapped by generating 9mer and/or 10mer peptides overlapping by 8 or 9 aa, respectively. To assess specificity/cross-reactivity toward CCNA2, we generated an overlapping CCNA2 pepmix as well as panel of 15 peptides spanning regions within CCNA2 that were analogous to immunogenic CCNA1-derived peptides. Lyophilized peptides were reconstituted at 10 mg/mL in dimethyl sulfoxide (Sigma-Aldrich).
DC generation
Monocytes were isolated from PBMCs by plastic adherence and cultured in DC medium (CellGenix USA, Portsmouth, NH) with 800 U/mL granulocyte macrophage-colony stimulating factor (GM-CSF) and 400 U/mL IL4 (R&D Systems) for 5 days. IL-4 and GM-CSF were replenished on day 3. On day 5, DCs were matured in DC medium supplemented with 1 µg/mL prostaglandin E2 (Sigma-Aldrich), 10 ng/mL IL-1β, 10 ng/mL tumor necrosis factor α (TNF-α), 100 ng/mL IL-6, 800 U/mL GM-CSF, and 400 U/mL IL-4 (all from R&D Systems).
CCNA1-specific T-cell generation
Mature DCs were pelleted and pulsed for 30 to 60 minutes at 37°C with peptides, individually or pooled (50 ng/peptide). PBMCs were stimulated with CCNA1-peptide–pulsed DCs (PBMC/DC ratio of 10:1). Cells were cultured at 1 × 106/mL in T-cell medium supplemented with IL-7 (10 ng/mL), IL-12 (10 ng/mL), IL-15 (5 ng/mL) (all from Peprotech, Rocky Hill, NJ), and IL-6 (100 ng/mL). Cultures were fed between days 6 to 8 and split 1:1 if confluent. On day 9, T cells were harvested, counted using trypan blue to distinguish live and dead cells, resuspended at 0.5 × 106 cells/mL in T-cell medium supplemented with IL-15 and IL-7, and then restimulated with CCNA1-peptide–loaded DCs (10:1). After 3 or 4 days, cultures were fed with fresh medium supplemented with IL-15, and from day 7 after the second stimulation, cells were used for phenotypic and functional studies.
Flow cytometry
CCNA1 detection
Tumor cell lines were fixed and permeabilized with BD Cytofix/Cytoperm solution (BD Biosciences, San Jose, CA) for 15 minutes, then washed with phosphate-buffered saline (PBS) (Sigma-Aldrich), and incubated with anti-CCNA1 rabbit polyclonal immunoglobulin G antibody (ab53699, Abcam) for 30 minutes at 4°C in the dark. After further washing, cells were incubated with brilliant violet 421–conjugated donkey anti-rabbit antibodies (BioLegend, San Diego, CA) for 1 hour prior to analysis using Gallios flow cytometer.
Immunophenotyping
CCNA1-specific T cells were surface stained with monoclonal antibodies to CD3, CD4, CD8, CD56, CD62L, and CD45RO (Becton Dickinson, Pasadena, CA). For surface staining, cells were washed with PBS, pelleted, and antibodies added in saturating amounts (2-5 μL). After a 15-minute incubation at 4°C in the dark, cells were washed and analyzed. Approximately 30 000 live cells were acquired using Gallios, and data were analyzed using Kaluza software (Version 1.3, Beckman Coulter).
Functional studies
ELISpot
The enzyme-linked immunospot (ELISpot) assay was used to quantitate the frequency of antigen-specific interferon γ (IFN-γ)– and granzyme B–secreting T cells. T-cell populations were resuspended at 2 × 106/mL in T-cell medium, and 100 μL was added to each ELISpot well. Antigen-specific activity was measured after direct peptide/pepmix exposure, with PHA (1 μg/mL) and unstimulated cells serving as positive and negative controls, respectively. After overnight incubation, plates were developed as per manufacturer instructions, dried at room temperature, and sent to ZellNet Consulting (New York, NY) for quantification.
ICS
To measure polyfunctionality and determine whether CCNA1 specificity was detectable in the CD4 or CD8 populations, intracellular cytokine staining (ICS) was performed. Briefly, cells resuspended at 2 × 106/mL in T-cell medium and then stimulated with test or control pepmix in the presence of CD28 and CD49d (1 µg/mL) (BD Biosciences), followed by the addition of BD GolgiStop and BD GolgiPlug, which contains monensin and brefeldin A, respectively. After an overnight incubation, T cells were washed with PBS, pelleted, and surface stained with CD8 and CD3, and after a 15-minute incubation at 4°C, the cells were washed, pelleted, fixed, and permeabilized with Cytofix/Cytoperm Solution (BD Biosciences). Each well was washed with BD Perm/Wash Buffer (1X) prior to staining with IFN-γ and TNF-α antibodies (BD Biosciences). Subsequently, cells were washed again, and at least 75 000 live cells were acquired using a Gallios flow cytometer. Analysis was performed using Kaluza software.
Cr51 release assay
The cytolytic potential of CCNA1-specific T cells was assessed using a 4- to 6-hour Cr51 release assays. The targets were chromium (Cr51)-labeled peptide-loaded autologous and partially HLA-matched PHA blasts and tumor cell lines. Unpulsed PHA blasts were used as a negative control. The percentage of specific lysis was calculated as [(experimental release − spontaneous release)/(maximum release − spontaneous release)] × 100. For HLA-blocking studies, labeled targets were preincubated with major histocompatibility complex (MHC) class I or II antibodies (Santa Cruz Biotechnology, Santa Cruz, CA) for 30 minutes at 37°C prior to the addition of effector T cells.
Results
CCNA1-specific T cells in healthy donors
To investigate the immunogenicity of CCNA1, we exposed PBMCs from healthy donors (n = 12) with diverse HLA types (supplemental Table 1) to pepmix-loaded DCs followed by expansion in T-cell medium supplemented with Th1 polarizing, proproliferative, and prosurvival cytokines. After 16 days and 2 rounds of in vitro stimulation, we achieved a mean 5.1- ± 0.6-fold increase in cell numbers (Figure 1A). The bulk cultures comprised predominantly of CD3+ T cells (81.4% ± 3.8%) representing CD8+ (cytotoxic: 64% ± 5%) and CD4+ (helper: 23% ± 4%) subsets that included cells showing both central (CD45RO+/62L+: 29% ± 7%) and effector (CD45RO+/62L−: 28% ± 6%) memory profile (Figure 1B; supplemental Figure 1), a composition that allows for immediate effector function and long-term in vivo persistence.17 To confirm that the expanded cultures were antigen specific and produced effector cytokines, we performed an IFN-γ ELISpot assay and determined that all but 1 of the expanded lines exhibited anti-CCNA1 activity (detection of >30 spot forming colonies [SFC]/1 × 105 input cells), with a mean frequency of 377 ± 98 SFC/1 × 105 input cells (median, 345; range, 38-1188 in responders) with no nonspecific activity (control: mean 2 ± 0 SFC/1 × 105 cells, P < .05; Figure 1C). These data confirm that CCNA1 can elicit T-cell responses in the majority of healthy donors, irrespective of HLA type.
![CCNA1-specific T-cell expansion and characterization. (A) Fold expansion of CCNA1-specific T cells (mean ± standard error of the mean [SEM]), based on cell counting using trypan blue exclusion (n = 12). (B) Immunophenotype of the expanded cell lines as assessed by flow cytometric analysis. (C) Specificity of expanded T cells as determined by IFN-γ ELISpot assay and data are presented as SFC/1 × 105 input cells. The whiskers of the plot indicate minimum and maximum values; boxes indicate median and interquartile ranges.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/2/10.1182_bloodadvances.2019000715/3/m_advancesadv2019000715f1.png?Expires=1769136462&Signature=s8Gucu2slluEJt0jmNcqnhxRXe-hgdbLscQU5H4xWfYH2Fk2OiZ6~nyItigLEa-jF1-hwNPouolWbILVJW~CzW7A~XZW682Oj2~xvnRw~2r98DU-dkuQ-1CMpCYFfki6JFZdaGZupkyq6yOCHt1-Oq5az~T9eCK75vqSjtpBW3jtN6NsqrJZPBX66hJa94P5O4jOM3MBZKnRB0J0U71s9I~dIwoXhFZVIP7xpHm7yPHMdsiYGk7icLl8VkqHlcPDMX44u2wl2JWdkTKyFLXSwFwvpHKPRKSzreExxm7rZGX5xoxHopPvZ7cFxdBBxy-1cC8VvdQbT1alh7GwOAM-Zg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CCNA1-specific T-cell expansion and characterization. (A) Fold expansion of CCNA1-specific T cells (mean ± standard error of the mean [SEM]), based on cell counting using trypan blue exclusion (n = 12). (B) Immunophenotype of the expanded cell lines as assessed by flow cytometric analysis. (C) Specificity of expanded T cells as determined by IFN-γ ELISpot assay and data are presented as SFC/1 × 105 input cells. The whiskers of the plot indicate minimum and maximum values; boxes indicate median and interquartile ranges.
CCNA1-specific T-cell expansion and characterization. (A) Fold expansion of CCNA1-specific T cells (mean ± standard error of the mean [SEM]), based on cell counting using trypan blue exclusion (n = 12). (B) Immunophenotype of the expanded cell lines as assessed by flow cytometric analysis. (C) Specificity of expanded T cells as determined by IFN-γ ELISpot assay and data are presented as SFC/1 × 105 input cells. The whiskers of the plot indicate minimum and maximum values; boxes indicate median and interquartile ranges.
Ex vivo–expanded CCNA1-specific T cells are polyfunctional
We next examined whether both CD4+ and CD8+ T cells contained CCNA1-reactive cells by performing ICS for IFN-γ. Figure 2A shows representative results from 1 donor (left) and summary data for all 11 responding donors (right), demonstrating that the dominant CCNA1-specific activity was detected in the CD8+ compartment (14.6% ± 1.4%, n = 11) with weaker CD4-mediated reactivity (4.8% ± 0.46%). In addition to IFN-γ, the expanded cells also produced TNF-α (Figure 2B, left, representative data; right, mean dual IFN-γ/TNF-α cytokine-producing cells in all responding donors [CD8+: 4.7% ± 0.1%, CD4: 0.6% ± <0.01%; n = 11]), confirming that these CCNA1-reactive cells are Th1 polarized and produce multiple effector cytokines upon antigen exposure.
![CCNA1-specific T cells are polyfunctional and cytotoxic. (A) Representative IFN-γ ICS data from 1 donor (left), and summary data for all responding donors (right; mean ± standard error of the mean; n = 11). (B) ICS analysis of dual (IFN-γ and TNF-α) cytokine secreting cells (representative data, left; summary data [n = 11], right). (C) ICS analysis of donor 8’s cell line gated on CD8+ T cells, showing CCNA1 specificity in the CD8 compartment. (D) These specific cells lysed autologous CCNA1 peptide-pulsed PHA blasts in a MHC class I–restricted manner, as assessed in a 4-hour Cr51 cytotoxicity assay at a range of effector/target (E:T) ratios. (E) Cytotoxic activity from 2 additional donor cell lines against autologous pulsed and unpulsed PHA blasts at an E:T 20:1 ratio.](https://ash.silverchair-cdn.com/ash/content_public/journal/bloodadvances/4/2/10.1182_bloodadvances.2019000715/3/m_advancesadv2019000715f2.png?Expires=1769136462&Signature=WeKg9m3M8b47oWAG8qXuQ45QpI8-uwvyW~y~y~8cKbiiuKB0o5v5Z379Jb3xsv5AyAVhppf-WSH0wmizQgot38TcqNmU3vpVRnsuApVHgxcogBI2sOT7U60Q2~yqC5K5UfZqzO1VNQujLJCkwzWS0lMgK04ftVoVDkDRF568UYWVSxjxw5w7qnKb0rKGkRVjVXufupMPpZRg4nT1JPXIenNaHIUZeXs2vxn-5WPeZmrGUMbBSGYERjku7mSxO9GmWqqSBthDEXf1dC98m04noDMks6HRsCqzoKp5hr6YK3ZP0CZPSQLB5-NvQ0BBpCfOI-VCqThDY5zzwCKKtVhKlg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CCNA1-specific T cells are polyfunctional and cytotoxic. (A) Representative IFN-γ ICS data from 1 donor (left), and summary data for all responding donors (right; mean ± standard error of the mean; n = 11). (B) ICS analysis of dual (IFN-γ and TNF-α) cytokine secreting cells (representative data, left; summary data [n = 11], right). (C) ICS analysis of donor 8’s cell line gated on CD8+ T cells, showing CCNA1 specificity in the CD8 compartment. (D) These specific cells lysed autologous CCNA1 peptide-pulsed PHA blasts in a MHC class I–restricted manner, as assessed in a 4-hour Cr51 cytotoxicity assay at a range of effector/target (E:T) ratios. (E) Cytotoxic activity from 2 additional donor cell lines against autologous pulsed and unpulsed PHA blasts at an E:T 20:1 ratio.
CCNA1-specific T cells are polyfunctional and cytotoxic. (A) Representative IFN-γ ICS data from 1 donor (left), and summary data for all responding donors (right; mean ± standard error of the mean; n = 11). (B) ICS analysis of dual (IFN-γ and TNF-α) cytokine secreting cells (representative data, left; summary data [n = 11], right). (C) ICS analysis of donor 8’s cell line gated on CD8+ T cells, showing CCNA1 specificity in the CD8 compartment. (D) These specific cells lysed autologous CCNA1 peptide-pulsed PHA blasts in a MHC class I–restricted manner, as assessed in a 4-hour Cr51 cytotoxicity assay at a range of effector/target (E:T) ratios. (E) Cytotoxic activity from 2 additional donor cell lines against autologous pulsed and unpulsed PHA blasts at an E:T 20:1 ratio.
Next, to examine their cytolytic potential, we incubated expanded CCNA1-specific T cells with autologous PHA blasts (control), CCNA1 pepmix-pulsed PHA blasts alone or in the presence of MHC class I or HLA-DR blocking antibodies. Figure 2C-D shows representative results from donor 8, whose expanded CCNA1-specific T cell line was predominantly CD8+ (89%, not shown) T cells with a high frequency of CD8+ CCNA1-directed activity, as demonstrated by ICS (20.3% vs 0.04%, CCNA1 pepmix vs irrelevant pepmix; Figure 2C). As shown in Figure 2D, these cells were able to specifically lyse autologous CCNA1-loaded PHA blasts (40% specific lysis, 40:1 E:T), which could be diminished by blocking MHC class I (60% reduction in cytolytic effects, 40:1 E:T), while blocking MHC class II had no effect. These results were confirmed in 2 additional donors (20:1 E:T; Figure 2E), demonstrating that CCNA1-specific T cells can mediate cytolytic effects in vitro.
Identification of immunogenic CCNA1 epitopes and HLA-restricting alleles
We next assessed the breadth of epitope specificity within CCNA1 by measuring responses to all 102 15mers spanning the entire antigen, which were arranged into 21 minipools such that each peptide was represented in 2 minipools. Figure 3A shows results from donor 2, whose line demonstrated the highest specificity for minipools 1, 3, 4, and 16, which intersected to identify 3 potentially stimulatory 15mers (45: aa 177-191, EAEIRHRPKAHYMKK; 47: aa 185-199, KAHYMKKQPDITEGM; and 48: aa 189-203, MKKQPDITEGMRTIL) (Figure 3B). To identify the stimulating peptide, we exposed the T cells to each individual peptide and identified 48 as immunogenic to CD8+ T cells (Figure 3C-D). Finally, to identify the HLA-restricting allele, we used autologous (HLA-A24, 29; B7, 35) and partially HLA matched peptide-pulsed PHA blasts as targets (Figure 3E). T cells reactive against peptide 48 recognized autologous and HLA-B7–matched allogeneic targets, confirming that CCNA1-directed activity in this line was mediated by CD8+ T cells specific for an HLA-B7–restricted epitope. Finally, to identify the minimal epitope, we used a panel of 9mers overlapping by 8 aa spanning peptide 48, which revealed the 9mer (QPDITEGMR) as the minimal epitope. Supplemental Figure 2 shows similar results for donor 1, whose CCNA1 response mapped to peptide 50 (aa 197-211, EGMRTILVDWLVEVG), and proved to be HLA-B35 restricted. Table 1 summarizes all minimal epitopes with HLA restrictions that have been mapped to date. Supplemental Table 2 lists other immunogenic 15mers identified.
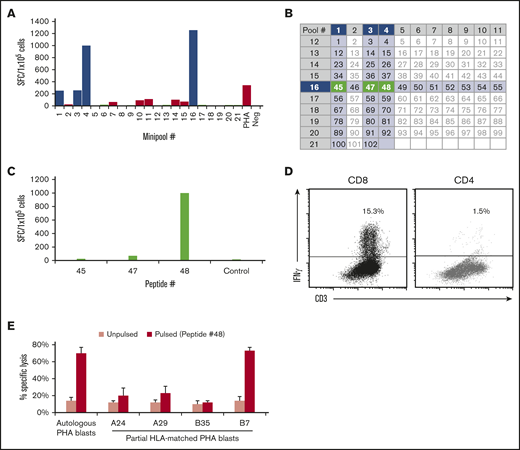
Identification of immunogenic CCNA1 epitopes and HLA-restriction profiling studies. (A) Donor 2’s cell line was tested against each of the 21 minipools by IFN-γ ELISpot (SFC/1 × 105). (B) Arrangement of peptides into minipools; donor 2’s reactive minipools (intersecting at peptides 45, 47, and 48) are shaded. Individual testing of these peptides identified peptide 48 as the stimulatory epitope (C), which was then confirmed by IFN-γ ICS (D). (E) Donor 2’s cell line lysed peptide-pulsed autologous and allogeneic targets matched at HLA-B7 but not at other alleles in a 4-hour Cr51 cytotoxicity assay, thus determining that peptide 48 is presented in context of HLA-B7. Results are presented as percent specific lysis, E:T 40:1.
Identification of immunogenic CCNA1 epitopes and HLA-restriction profiling studies. (A) Donor 2’s cell line was tested against each of the 21 minipools by IFN-γ ELISpot (SFC/1 × 105). (B) Arrangement of peptides into minipools; donor 2’s reactive minipools (intersecting at peptides 45, 47, and 48) are shaded. Individual testing of these peptides identified peptide 48 as the stimulatory epitope (C), which was then confirmed by IFN-γ ICS (D). (E) Donor 2’s cell line lysed peptide-pulsed autologous and allogeneic targets matched at HLA-B7 but not at other alleles in a 4-hour Cr51 cytotoxicity assay, thus determining that peptide 48 is presented in context of HLA-B7. Results are presented as percent specific lysis, E:T 40:1.
Summary of immunogenic CCNA1 sequences, with associated CD4 or CD8 response, HLA restriction, and minimal epitopes
| Peptide | Peptide sequence | Position | Response | HLA restriction | Minimal epitope |
|---|---|---|---|---|---|
| 48 | MKKQPDITEGMRTIL | 189-203 | CD8 | B7 | QPDITEGMR |
| 50 | EGMRTILVDWLVEVG | 197-211 | CD8 | B35 | MRTILVDWL |
| 50 | EGMRTILVDWLVEVG | CD8 | A2 | RTILVDWLV | |
| 55 | RAETLYLAVNFLDRF | 217-231 | CD8 | B35 | YLAVNFLDR |
| 55 | RAETLYLAVNFLDRF | CD8 | A2 | AETLYLAVN | |
| 82 | AELSLLEADPFLKYL | 325-339 | CD8 | A2 | LLEADPFLK |
| 82 | AELSLLEADPFLKYL | CD8 | A2 | EADPFLKYL | |
| 85 | KYLPSLIAAAAFCLA | 337-351 | CD8 | A2 | SLIAAAAFCLA |
| Peptide | Peptide sequence | Position | Response | HLA restriction | Minimal epitope |
|---|---|---|---|---|---|
| 48 | MKKQPDITEGMRTIL | 189-203 | CD8 | B7 | QPDITEGMR |
| 50 | EGMRTILVDWLVEVG | 197-211 | CD8 | B35 | MRTILVDWL |
| 50 | EGMRTILVDWLVEVG | CD8 | A2 | RTILVDWLV | |
| 55 | RAETLYLAVNFLDRF | 217-231 | CD8 | B35 | YLAVNFLDR |
| 55 | RAETLYLAVNFLDRF | CD8 | A2 | AETLYLAVN | |
| 82 | AELSLLEADPFLKYL | 325-339 | CD8 | A2 | LLEADPFLK |
| 82 | AELSLLEADPFLKYL | CD8 | A2 | EADPFLKYL | |
| 85 | KYLPSLIAAAAFCLA | 337-351 | CD8 | A2 | SLIAAAAFCLA |
CCNA1-specific T cells kill CCNA1-expressing tumors
To assess whether CCNA1-activated T-cell lines could mediate antitumor effects, we cocultured donor 2’s T-cell line (with HLA-B7–restricted activity) with the tumor cell lines U266 (HLA-B7+/CCNA1+), KG1a (HLA-B7−/CCNA1+), U937 (HLA-B7−/CCNA1weak+), and autologous PHA blasts (HLA-B7+/CCNA1−) (supplemental Figure 3 shows CCNA1 expression in tumor cell lines). As shown in Figure 4A, only U266 was killed (53% specific lysis, E:T 20:1), with minimal activity against the other targets (KG1a, 3%; U937, 12%; PHA blasts, 3%). These results were supported by IFN-γ and granzyme B ELISpot (Figure 4B). In addition, we generated CCNA1-specific T cell lines from 3 additional donors with HLA-restricted CCNA1 reactivity (Figure 4C), which were cocultured (E:T 40:1) with a panel of partially HLA-matched (±CCNA1+) leukemic cell lines. Autologous PHA blasts (±peptide) served as positive and negative controls, respectively. Only cell lines that were matched at relevant HLA alleles and endogenously expressing CCNA1 were killed with minimal activity against other targets. We further extended our studies to the assessment of CCNA1 T-cell activity against partially HLA-matched primary leukemic cells. Figure 4D shows that donor 8’s CCNA1-specific cell line with confirmed HLA-A2–restricted activity specifically killed an A2+/CCNA1+ AML sample (from patient 1) with no activity against an A2−/CCNA1+ primary leukemic sample from patient 2.
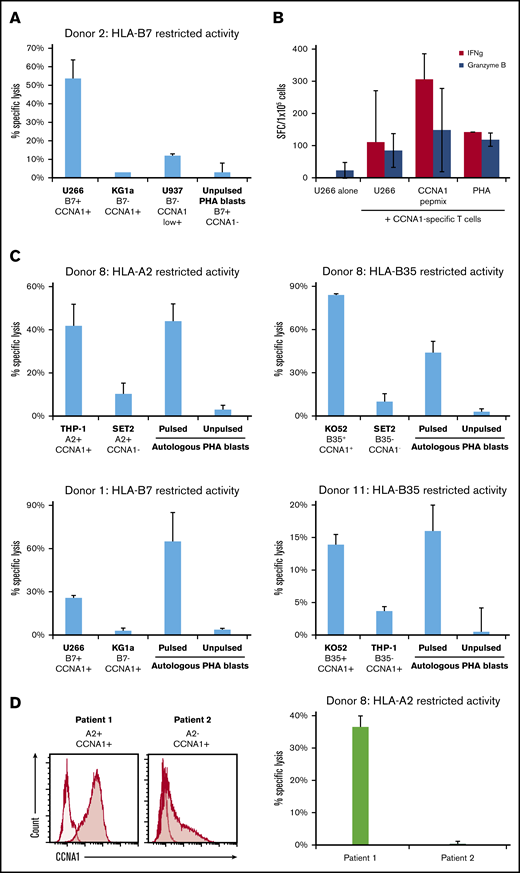
CCNA1-specific T cells show cytotoxicity against HLA-matched, CCNA1+tumor cell lines. (A) Donor 2’s cell line showed high specific lysis toward U266 (HLA-B7+ and CCNA1+) in a 4-hour Cr51-release assay, compared with targets that express either HLA-B7 or CCNA1 alone. (B) Similarly, donor 2’s cell line produced IFN-γ and granzyme B, as assessed by ELISpot, following incubation with the U266 cell line, but not when U266 was cultured alone. (C) Further examples of CCNA1-specifc T cells from donors with characterized HLA-restricted CCNA1 reactivity, showing specific lysis toward CCNA1-expressing leukemic cell lines matched at the relevant HLA allele, with minimal activity against targets that were HLA mismatched and/or CCNA1 negative. (D) CCNA1 expression of 2 primary AML cell lines (left; CCNA1, shaded curve; no primary control, unshaded curve) with HLA-A2 status of patients, and specific lysis of patient 1’s AML blasts (right).
CCNA1-specific T cells show cytotoxicity against HLA-matched, CCNA1+tumor cell lines. (A) Donor 2’s cell line showed high specific lysis toward U266 (HLA-B7+ and CCNA1+) in a 4-hour Cr51-release assay, compared with targets that express either HLA-B7 or CCNA1 alone. (B) Similarly, donor 2’s cell line produced IFN-γ and granzyme B, as assessed by ELISpot, following incubation with the U266 cell line, but not when U266 was cultured alone. (C) Further examples of CCNA1-specifc T cells from donors with characterized HLA-restricted CCNA1 reactivity, showing specific lysis toward CCNA1-expressing leukemic cell lines matched at the relevant HLA allele, with minimal activity against targets that were HLA mismatched and/or CCNA1 negative. (D) CCNA1 expression of 2 primary AML cell lines (left; CCNA1, shaded curve; no primary control, unshaded curve) with HLA-A2 status of patients, and specific lysis of patient 1’s AML blasts (right).
Examining the safety potential of CCNA1-specific T cells
Cyclins are a group of related proteins involved in cell cycle regulation. There are 2 members of cyclin A: CCNA1, which is expressed during meiosis, with high levels of tissue expression restricted to the testes, and as such is a CTA; and CCNA2, which shares 56% sequence identity with CCNA1 and is expressed in dividing somatic cells. Hence, one of the concerns in targeting CCNA1 is the potential for cross-reactive recognition of CCNA2-expressing cells, leading to a risk of “off tumor” effects. To address this potential concern, we generated an overlapping peptide library spanning CCNA2. We then exposed CCNA1-reactive T cells to either the CCNA1 or CCNA2 peptide libraries and found no cross-reactivity (n = 6, 1317 ± 344 vs 1.8 ± 0.8 SFC/1 × 105) (Figure 5A). Furthermore, we generated a series of peptides encoding CCNA2-derived sequences that overlapped with immunogenic CCNA1 regions (Table 2) and exposed CCNA1-reactive T cells to either the CCNA1 or CCNA2 peptide equivalents. Figure 5B shows representative results from 2 donors, while Figure 5C summarizes data from 13 lines with specificity for 5 distinct CCNA1 epitopes. As shown in Figure 5B, donor 1’s T-cell line demonstrated specificity for peptide 50 with no activity to the CCNA2 counterpart (612 ± 114 vs 8.3 ± 0.9 SFC/1 × 105, respectively). Similarly, donor 2’s T-cell lines with specificity for peptide 48 had no recognition of the analogous sequence in CCNA2 (322 ± 31 vs 0.5 ± 0.5 SFC/ 1 × 105, respectively). Taken together, these data demonstrate the exquisite specificity of CCNA1-specific T-cell lines.
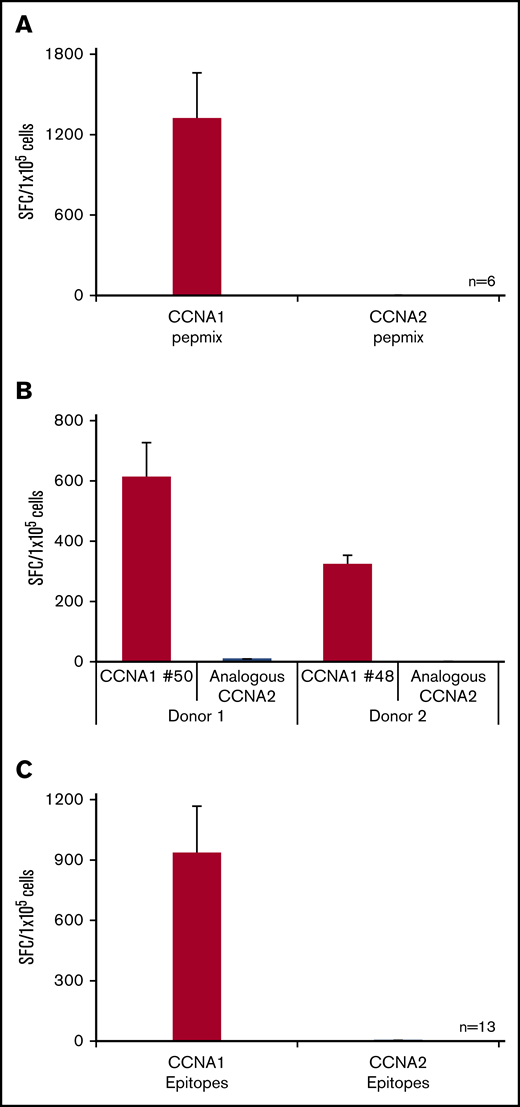
CCNA1-specific T cells do not cross-reactively recognize CCNA2. (A) IFN-γ ELISpot results of 6 CCNA1-directed T cell lines following exposure to either CCNA1 or CCNA2 pepmix. (B) Two representative cell lines following exposure to either CCNA1 epitopes or analogous peptides from CCNA2. (C) Summary data (n = 13).
CCNA1-specific T cells do not cross-reactively recognize CCNA2. (A) IFN-γ ELISpot results of 6 CCNA1-directed T cell lines following exposure to either CCNA1 or CCNA2 pepmix. (B) Two representative cell lines following exposure to either CCNA1 epitopes or analogous peptides from CCNA2. (C) Summary data (n = 13).
Immunogenic CCNA1 sequences and corresponding analogous CCNA2 peptides
| Peptide | CCNA1 sequence | Analogous CCNA2 sequence |
|---|---|---|
| 48 | MKKQPDITEGMRTIL | MKKQPDITNSMRAIL |
| 50 | EGMRTILVDWLVEVG | NSMRAILVDWLVEVG |
| 55 | RAETLYLAVNFLDRF | QNETLHLAVNYIDRF |
| 68 | YITDDTYTKRQLLKM | YITDDTYTKKQVLRM |
| 82 | AELSLLEADPFLKYL | GELSLIDADPYLKYL |
| 85 | KYLPSLIAAAAFCLA | KYLPSVIAGAAFHLA |
| Peptide | CCNA1 sequence | Analogous CCNA2 sequence |
|---|---|---|
| 48 | MKKQPDITEGMRTIL | MKKQPDITNSMRAIL |
| 50 | EGMRTILVDWLVEVG | NSMRAILVDWLVEVG |
| 55 | RAETLYLAVNFLDRF | QNETLHLAVNYIDRF |
| 68 | YITDDTYTKRQLLKM | YITDDTYTKKQVLRM |
| 82 | AELSLLEADPFLKYL | GELSLIDADPYLKYL |
| 85 | KYLPSLIAAAAFCLA | KYLPSVIAGAAFHLA |
Detection of CCNA1-specific T cells in AML patients
Based on our in vitro studies, we hypothesized that individuals with AML who had received an allo-HSCT and were in remission might have circulating (endogenous) CCNA1-reactive cells capable of mediating protective antitumor effects. To test this hypothesis, we stimulated patient-derived PBMCs (n = 8) with CCNA1 using samples that were collected at 2 or 3 months posttransplant while all were in remission (supplemental Table 2 summarizes patient characteristics). We then followed these patients for clinical outcomes and found a direct correlation between the presence and magnitude of CCNA1-directed T cells measured early posttransplant and risk of disease relapse by 1 year post-HSCT. Figure 6 shows the T-cell responses, assessed by IFN-γ ELISpot in relapse-free patients (n = 4) compared with those who subsequently relapsed (n = 4) (mean 171 vs 7.13 SFC/1 × 105, P = .03). While the sample size is small, these findings suggest the protective benefit conferred by endogenous CCNA1-specific T cells post-HSCT.
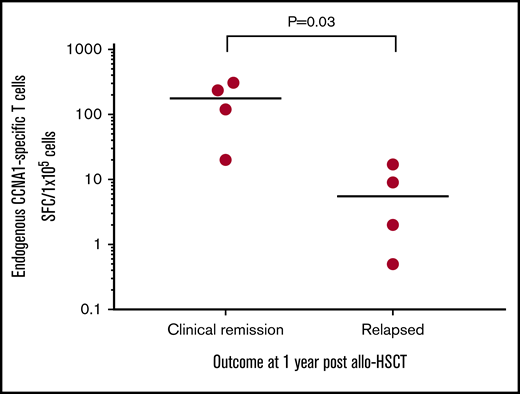
Endogenous CCNA1-specific T cells detected in AML patients who remain in clinical remission posttransplant. CCNA1-specific T-cell responses detected in PBMCs (by IFN-γ ELISpot) isolated from patients with AML who received allo-HSCT (n = 8). Results are presented as SFC/1 × 105.
Endogenous CCNA1-specific T cells detected in AML patients who remain in clinical remission posttransplant. CCNA1-specific T-cell responses detected in PBMCs (by IFN-γ ELISpot) isolated from patients with AML who received allo-HSCT (n = 8). Results are presented as SFC/1 × 105.
Discussion
In this study, we sought to assess the potential of CCNA1 as an immunotherapeutic target for the treatment of AML, the most common acute leukemia in adults with a 5 year OS of just 28.3%.1 In peripheral blood samples collected from 12 healthy donors of diverse ethnic backgrounds (as reflected by their HLA profiles), we identified CCNA1-directed T-cell activity (defined as SFC >30/1 × 105 input cells) in 92% of individuals screened (n = 11/12), attesting to the immunogenic nature of this antigen. Reactive cells, which were Th1 polarized, polyfunctional, and cytotoxic, could be readily expanded with repetitive rounds of in vitro stimulation, hence supporting the feasibility of producing such cells in clinically relevant numbers for adoptive transfer. Furthermore, in AML patients, we found a correlation between detection of endogenous CCNA1 reactivity and prolonged disease remission, suggestive of the protective antitumor effects mediated by such cells. Finally, given that CCNA1 belongs to a highly conserved family of cyclin proteins (4 main classes in humans: CCNA, CCNB, CCND, and CCNE; range, 25% to 56% homology) that are ubiquitously expressed and play a central role in cell cycle regulation,18,19 we sought to address the recognition profile of reactive cells. Of note, these ex vivo–expanded populations were exquisitely specific for CCNA1 and inert upon encounter with other cyclins, supporting the safety of our proposed immunotherapeutic approach. Taken together, these data support the development of immune-based approaches to target CCNA1 for the treatment of AML.
AML is an aggressive hematological malignancy characterized by the clonal expansion of myeloid progenitor cells, with consequential suppression of normal hematopoiesis resulting in symptoms of cytopenia, including tiredness, potentially life-threatening bleeding, and infections.20 It is a heterogeneous disease with respect to clinical presentation and pathologic features, including blast morphology, genetics (eg, FLT3-ITD and NPM1 mutations), and immunophenotypic expression of hematopoietic precursor (HLA-DR, CD34, and CD117) and differentiated myeloid markers (CD13 and CD33).20,21 Additionally, there is frequent aberrant protein expression of LAAs, including WT1, survivin, and PRAME.12,22
Treatment decisions are guided by patient age, comorbidities, performance status, and risk stratification based on genetic features.23,24 For example, ∼60% of newly diagnosed AML patients are deemed fit to tolerate intensive chemotherapy25 and receive induction with cytarabine and an anthracycline, which produces remission rates as high as 83%.26 For the ∼25% of patients with adverse-risk genetic features (eg, FLT3-ITD and complex karyotype)27 or those with relapsed/refractory disease, consolidation with allo-HSCT after remission offers the best chance of cure.28,29 However, the transplant procedure is highly toxic and associated with transplant-related mortality rates of 10% to 30%, while 50% of transplant recipients ultimately experience disease relapse that is associated with a dismal median survival of just 6 months.3 Recently approved agents for the treatment of relapsed/refractory AML include gemtuzumab ozogamicin, an anti-CD33 drug–linked monoclonal antibody, and the IDH1 and IDH2 inhibitors ivosidenib and enasidenib, respectively. However, these new therapies address only a subset of patients expressing the relevant drug targets, are associated with severe reactions (such as veno-occlusive disease in those treated with gemtuzumab ozogamicin and differentiation syndrome in those administered enasidenib), and produce clinical remissions in a minority, highlighting the need for novel therapies.30-32
The adoptive transfer of tumor-targeting T cells has proven effective in the treatment of various diseases, particularly for CD19-directed CAR T cells for B-cell acute lymphoblastic leukemia in children and young adults.6 However, this benefit must be weighed against the associated “on target, off tumor” effects; interaction of transgenic T cells with normal CD19+ B cells has resulted in lifelong B-cell aplasia in treated patients, though can be compensated by intravenous immunoglobulin replacement.33 Unfortunately, for AML, a “safe” tumor-expressed cell surface antigen has yet to be identified. For example, CD33 and CD123 are expressed in >80% of blasts but also on myeloid progenitor cells, whose eradication would be clinically intolerable.34,35 The alternative approach suggested in the current study entails the adoptive transfer of T cells whose native TCRs recognize aberrantly expressed endogenous proteins such as CCNA1, NY-ESO-1, and WT1. Indeed, Chapuis et al13 generated donor-derived HLA-A*0201–restricted WT1-specific CD8 T cells by repetitive stimulation of PBMCs using peptide-pulsed DCs and upon the transfer of up to 1 × 1010 cells/m2 saw no toxicities or graft-versus-host disease. Furthermore, the cells produced antileukemic responses in 2 of 11 patients (in 1 patient, there was a reduction of blasts from 7% to 0%, with subsequent relapse following the disappearance of the infused T cells; the other patient with minimal residual disease at the time of infusion remained in remission 19 months after T cells, with normalization of cytogenetics). While this study demonstrates the protective potential of targeting aberrantly expressed endogenous antigens such as CCNA1, ultimately, the therapeutic benefit of an adoptive immunotherapeutic approach will likely be maximized by simultaneously targeting multiple leukemia-expressed antigens.36,37
The cyclins are a family of proteins that regulate the cell cycle.19 Each family contains subfamily members (eg, CCNA1 and CCNA2 fall within the CCNA family and share 56% sequence identity).18 Both CCNA1 and CCNA2 couple with cyclin-dependent kinases (CDK1 and CDK2) to mediate the G1/S and G2/M phase transition.38,39 While CCNA2 is ubiquitously expressed in all dividing somatic cells, CCNA1 is primarily involved in spermatogenesis; thus, high tissue CCNA1 expression is restricted to spermatocytes in the testes.40,41
Given its upregulation in both cell lines and AML patient samples, various groups have explored the role of CCNA1 in tumorigenesis. For instance, Ji et al42 reported that the CCNA1-CDK2 complex mediates oncogenesis through inhibition of apoptosis by phosphorylating retinoblastoma protein. Krug et al43 found that CCNA1 overexpression repressed WT1, a tumor suppressor gene, at both the messenger RNA and protein levels, which led to abrogation of the G1 cell cycle arrest. CCNA1 also interacts directly with B-myc (a transcription factor involved in proliferation), which can further transactivate the CCNA1 promoter to set up an autoregulatory feedback loop.44 In transgenic mouse models, overexpression of CCNA1 in the myeloid lineage can induce abnormal myelopoiesis with increased premature cells in the bone marrow and transformation to overt leukemia in ∼15%.45 In AML, CCNA1 is overexpressed in >80% of malignant blasts14 as well as leukemic stem cells, as reported by Oshsenreither et al.15 Thus, immunotherapeutic targeting should facilitate not only tumor debulking but also the elimination of the stem cell population implicated in chemoresistance and relapse. Beyond AML, CCNA1 has been reported to be upregulated in multiple cancers, including testicular germ cell tumors,46 ovarian cancer,47 and esophageal cancer,48 supporting the potential for broad applicability of adoptively transferred CCNA1-specific T cells.
Given the homology between CCNA1 and CCNA218 and that CCNA2 is expressed in a range of normal tissues (eg, lymph nodes, gastrointestinal and urinary tract, and lungs49 ), we sought to comprehensively examine the potential for cross-reactive T-cell recognition that might lead to multiorgan damage. Indeed, such events have been previously reported with TCR-engineered T cells directed at MAGE-A3, a CTA overexpressed by a range of solid tumors. Linette and colleagues50 administered autologous affinity-enhanced HLA-A1*01–restricted TCR-transduced T cells directed towards the MAGE-A3 epitope EVDPIGHLY (MAGE-A3a3a T cells), to treat MAGE-A3+ metastatic melanoma and myeloma. The first 2 patients were infused with 5.3 × 109 and 2.4 × 109 MAGE-A3a3a T cells, respectively, and experienced severe acute cardiac toxicities resulting in their deaths within 5 days of infusion. The autopsies revealed myocyte necrosis and marked CD3 infiltration, likely due to transgenic T-cell recognition of a similar epitope derived from the muscle protein titin (ESDPIVAQY) expressed on myocytes.50 Of note, MAGE-A3a3a TCR T cells showed >10-fold activation against HLA-A*01+/titin+/MAGE-A3− beating myocytes compared with the native MAGE-A3 TCR. This highlights the danger of T-cell recognition of off-target epitopes, particularly in the context of using affinity-enhanced TCRs. Our approach utilizes endogenous (nonengineered) T cells, which we extensively analyzed to assess the potential for cross-reactivity, and our CCNA1-specific cell lines were inert when cocultured with CCNA2 peptides, thereby supporting their in vivo safety. Furthermore, we detected endogenous CCNA1-reactive T cells in AML patients in prolonged remission after allo-HSCT, not only highlighting the safety of such reactive cells but also supporting the protective potential of such cells for future immunotherapeutic use.
In conclusion, our study supports CCNA1 as a suitable immunotherapeutic target for AML. CCNA1 is immunogenic, and it is feasible to generate donor-derived CCNA1-specific T cells that do not cross-react with similar/homologous proteins such as CCNA2 that are widely expressed in a range of tissues. In addition, we discovered an association between endogenous CCNA1-specific T cells in AML patients and ongoing clinical remission. These findings support targeting CCNA1 (in combination with other tumor-expressed antigens) in future clinical trials of adoptively transferred T cells for AML treatment.
Acknowledgments
The authors thank Walter Mejia for artwork and formatting of manuscript figures.
W.K.L. is supported by the Cancer Research Trust New Zealand, a Murray Jackson Clinical Fellowship, and a HSANZ New Investigator Scholarship. P.L. is supported by an ASH Scholar Award, an ASBMT New Investigator Award, a Leukemia Texas research grant, and an Edward P. Evans Foundation MDS Discovery research grant. A.M.L. is supported by Leukemia & Lymphoma Society Rising Tide Foundation (CCR-16-200), a Specialized Center of Research Award from the Leukemia & Lymphoma Society (7019-19), and a National Institutes of Health, National Cancer Institute SPORE in Lymphoma grant (P50 CA126752).
Authorship
Contribution: P.L., W.K.L., I.T., and A.M.L. conceived and designed the research; W.K.L., A.W., S.M., D.B., and P.L performed experiments; W.K.L., N.W., and P.L. analyzed the data; and W.K.L., A.M.L., and P.L wrote the manuscript.
Conflict-of-interest disclosure: A.M.L. is a cofounder and consultant/advisory board member for Allovir (an Elevatebio Company) and Marker Therapeutics and has ownership interest (including stock and patents) in both companies. The remaining authors declare no competing financial interests.
Correspondence: Premal Lulla, Texas Children’s Hospital and Houston Methodist Hospital, 1102 Bates Ave, Feigin Center 1780.06, Houston, TX 77030; e-mail: lulla@bcm.edu.

