

Key Points
PTPN22 deficiency promotes platelet aggregation, spreading, and clot retraction and accelerates arterial thrombus formation.
PTPN22 interacts with phosphorylated PDE5A (Ser92) and possesses intrinsic serine phosphatase activity in activated platelets.

Abstract
Protein tyrosine phosphatase nonreceptor type 22 (PTPN22) is a protein tyrosine phosphatase that negatively regulates T-cell signaling. However, whether it is expressed and functions in platelets remains unknown. Here we investigated the expression and role of PTPN22 in platelet function. We reported PTPN22 expression in both human and mouse platelets. Using PTPN22−/− mice, we showed that PTPN22 deficiency significantly shortened tail-bleeding time and accelerated arterial thrombus formation without affecting venous thrombosis and the coagulation factors VIII and IX. Consistently, PTPN22-deficient platelets exhibited enhanced platelet aggregation, granule secretion, calcium mobilization, lamellipodia formation, spreading, and clot retraction. Quantitative phosphoproteomic analysis revealed the significant difference of phosphodiesterase 5A (PDE5A) phosphorylation in PTPN22-deficient platelets compared with wild-type platelets after collagen-related peptide stimulation, which was confirmed by increased PDE5A phosphorylation (Ser92) in collagen-related peptide–treated PTPN22-deficient platelets, concomitant with reduced level and vasodilator-stimulated phosphoprotein phosphorylation (Ser157/239). In addition, PTPN22 interacted with phosphorylated PDE5A (Ser92) and dephosphorylated it in activated platelets. Moreover, purified PTPN22 but not the mutant form (C227S) possesses intrinsic serine phosphatase activity. Furthermore, inhibition of PTPN22 enhanced human platelet aggregation, spreading, clot retraction, and increased PDE5A phosphorylation (Ser92). In conclusion, our study shows a novel role of PTPN22 in platelet function and arterial thrombosis, identifying new potential targets for future prevention of thrombotic or cardiovascular diseases.
Introduction
Reversible protein tyrosine phosphorylation plays important roles in the modulation of multiple intracellular signaling events.1 In platelets, engagement of platelet surface receptors glycoprotein VI (GPVI) or GPIb-IX-IV by respective ligands collagen or von Willebrand factor exposed from subendothelial matrix at the site of vascular injury triggers the phosphorylation of several proteins (eg, Syk, phosphatidylinositol 3-kinase/AKT, phospholipase C γ2 [PLCγ2]), leading to activation and transduction of intraplatelet signaling pathway.2,3 Protein tyrosine phosphorylation is determined or tightly regulated by the dynamic balance between protein tyrosine kinases and protein tyrosine phosphatases (PTPs).4 In response to stimulus, protein tyrosine kinases phosphorylate signaling proteins to initiate platelet activation signals, whereas PTPs dephosphorylate the phosphorylated proteins to downregulate their function or activity to exert negative feedback regulatory effects on platelet function.4,5
PTPN22, the PTP, nonreceptor type 22, encodes a member of PTPs, lymphoid-specific tyrosine phosphatase in humans or proline, glutamic acid, serine, threonine-rich-domain phosphatase (PEP) in mice.6 PTPN22 is only expressed in hematopoietic cells and contains an N-terminal/PTP domain, interdomain, and C-terminal domain (mediates the binding to the Csk tyrosine kinase).7,8 The role and function of PTPN22 are best characterized in T cells, as it inhibits T-cell antigen receptor (TCR) signaling by dephosphorylating key kinases of the T cell–signaling pathway such as Lck, ZAP70, VaV, and EB1,9-11 which is further supported by increased TCR-mediated T-cell proliferation and expansion of the effector or memory T-cell compartment in PTPN22-deficient mice.12,13
Using transcriptome-based approaches, PTPN22 has been found to be expressed in megakaryocytes.14,15 However, whether platelets express PTPN22 remains unclear. In this study, we first showed that PTPN22 is expressed in both human and mouse platelets. Using PTPN22-deficient mice and PTPN22 inhibitor, we further characterized its role in platelet function and thrombosis. Our data showed a negative role of PTPN22 in platelet function and arterial thrombus formation.
Materials and methods
Animals
PTPN22−/− mice (C57BL/6) were generated by Cyagen Biosciences Inc. using CRISPR/Cas-mediated genome engineering. The mouse PTPN22 gene (GenBank accession number: NM_008979.2; Ensembl: ENSMUSG00000027843) is located on mouse chromosome 3 with 21 exons. Exon 3 to exon 11 and exon 2 to exon 13 were selected as target site (supplemental Figure 1, available on the Blood Web site). Two pairs of guide RNA targeting vectors were constructed and confirmed by sequencing. Cas9 messenger RNA and guide RNA generated by in vitro transcription were coinjected into fertilized eggs for knockout mouse production. The pups were genotyped by polymerase chain reaction followed by sequence analysis. All experimental procedures were approved by the Ethic Committee of Xuzhou Medical University.
PTPN22-deficient mice have been shown to display an accumulation of effector/memory T cells later in life with enhanced activities in response to TCR engagement.12 In addition, increased numbers of follicular helper T cells16 and expansion of regulatory T cells13 have also been observed. Moreover, these mice develop spontaneous germinal center formation, splenomegaly, and increased serum levels of immunoglobulin G1 (IgG1), IgG2a, and IgE.12 However, these mice do not display any clinical manifestations of autoimmunity. Interestingly, B-cell receptor signaling and B-cell development are normal in PTPN22-deficient mice.
Platelet preparation and analysis
Platelets were isolated as described previously.17,18 Platelet receptor expression, aggregation and activation, ultrastructure, spreading, clot retraction, in vitro thrombus formation under flow conditions, tail bleeding, and thrombus formation in vivo were performed as described elsewhere.19-23 Detailed methods are provided in the supplemental Methods.
Quantitative phosphoproteomics analysis
Platelets were treated with collagen-related peptide (CRP) (5 μg/mL) for 1 minute; they were then lysed in SDT (4% [w/v] sodium dodecyl sulfate [SDS], 100 mM Tris-HCl, 1 mM dithiothreitol, pH 7.6) buffer to extract protein, which was quantified by using a BCA Protein Assay Kit (Bio-Rad). Then, 20 µg of protein were separated on 12.5% SDS–polyacrylamide gel electrophoresis gel, and protein bands were visualized by Coomassie Blue R-250 staining followed by filter-aided sample preparation using 200 μg of proteins. Peptide mixture (100 μg) was labeled using TMT (Thermo Fisher Scientific) or iTRAQ (Applied Biosystems) reagent according to the protocols. Phosphopeptide enrichment (ie, immobilized metal ion affinity chromatography enrichment method) was performed by using High-Select Fe-NTA Phosphopeptide Enrichment Kit according to the manufacturer’s instructions (Thermo Scientific). After lyophilization, the phosphopeptides peptides were resuspended in 20 µL loading buffer (0.1% [v/v] formic acid) followed by liquid chromatography/tandem mass spectrometry analysis on a Q Exactive HF mass spectrometer (a hybrid-quadrupole/orbitrap mass spectrometer; Thermo Fisher Scientific) that was coupled to an EASY-nLC (Thermo Fisher Scientific) for 120 minutes. Tandem mass spectrometry spectra were searched by using MASCOT engine (Matrix Science; version 2.2) embedded into Proteome Discoverer 2.4 (Thermo Fisher Scientific).
Immunoprecipitation
After treatment with CRP (5 μg/mL), platelets were lysed in NP-40 lysis buffer for immunoprecipitation assay using antibody against PTPN22 or phosphodiesterase 5A (PDE5A; Ser92) as described previously.23
Recombinant protein expression and purification
Based on the structure (domains) of PTPN22, PTPN22 catalytic domain (PTPase), the catalytically inactive mutant form (C227S), or the trapping mutant form (D195A/C227S, DACS) was amplified and ligated into pGEX-4T-1 and then transformed into Escherichia coli. The glutathione S-transferase (GST) and GST-recombinant proteins were induced with 1 mM isopropyl-β-d-thiogalactopyranoside for 20 hours at 16°C followed by collection of the cells, which were then homogenized by sonication and centrifuged to obtain the supernatant. Protein in the supernatant was purified by using Glutathione Sepharose 4B (GE Healthcare), and the purity was >90% as evaluated by SDS–polyacrylamide gel electrophoresis.
In vitro PDE5A dephosphorylation assay
PTPN22-deficient platelets were stimulated with CRP (5 μg/mL) for 15 seconds and lysed, followed by incubation with GST-PTPN22 PTPase (in the presence or absence of 40 μM LTV-1), GST-PTPN22 C227S, or GST (0.4 μg) in HEPES buffer (50 mM HEPES, pH 7.1, 0.1 mM EDTA, 10 mM 2-mercaptoethanol) for 30 minutes at room temperature. PDE5A phosphorylation (Ser92) level was then detected by western blot. Phosphopeptide of PDE5A with Ser92 phosphorylation as a motif (GAPARKIpSASEFDR) or Syk with Tyr519/520 phosphorylation as a motif (ALRADENpYpYKAQTH) was synthesized and used as a substrate to measure the catalytic activity of PTPN22 on phosphorylated PDE5A (p-PDE5A) by using a Malachite Green assay (MilliporeSigma).
Statistical analysis
Data are shown as mean ± standard deviation or standard error where indicated and assessed by t test with one- or two-way analysis of variance. P < .05 indicates a significant difference.
Results
PTPN22 is expressed in platelets, and its deficiency enhances hemostasis and arterial thrombus formation in vivo
To evaluate whether platelets and megakaryocytes express PTPN22, we isolated human and mouse platelets or megakaryocytes. PTPN22 was detected in both human and mouse platelets or megakaryocytes using immunoblotting and immunofluorescence approaches (Figure 1A-B). The specificity of PTPN22 antibody was verified by using platelets isolated from PTPN22-deficient mice (Figure 1C). To evaluate whether PTPN22 deficiency alters the expression of other PTPNs, we measured PTPN1, PTPN6, and PTPN11 expression and found comparative expression levels between wild-type (WT) platelets and PTPN22-deficient platelets (supplemental Figure 2).
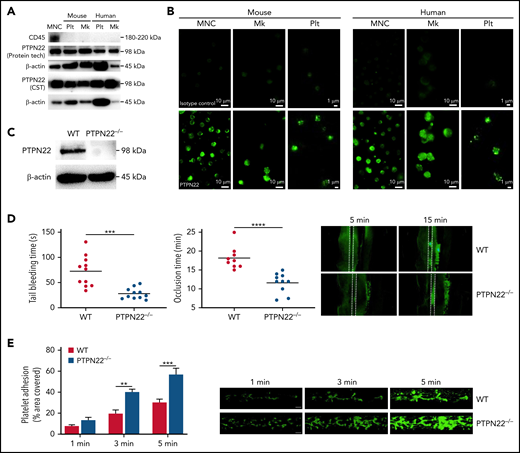
PTPN22 expression in platelets (Plt) and its role in hemostasis and thrombus formation. Human or mouse mononuclear cells (MNC) were isolated and used as positive control. Plt or megakaryocytes (Mk) from WT mouse or platelets from healthy individuals were isolated to measure PTPN22 expression by western blot with 2 different antibodies (A) or immunofluorescent staining (B). (C) PTPN22 expression in PTPN22−/− Plt was measured by western blot. Human megakaryocytic cell line (CMK) was used as human Mk. Data are representative of 3 independent experiments. Scar bar = 10 μm. (D) Tail bleeding time and FeCl3-induced arterial thrombosis in WT and PTPN22−/− mice (mean). Representative image of arterial thrombosis at different time point is shown. (E) Platelet adhesion on collagen under flow conditions. Whole blood was labeled with mepacrine and perfused through fibrillar collagen–coated BioFlux plates (Fluxion Biosciences) at 40 dynes/cm2 for 5 minutes. Platelet adhesion (covered area) was quantified at different time points (mean ± standard error, n = 6; one-way analysis of variance). **P < .01, ***P < .001, ****P < .0001.
PTPN22 expression in platelets (Plt) and its role in hemostasis and thrombus formation. Human or mouse mononuclear cells (MNC) were isolated and used as positive control. Plt or megakaryocytes (Mk) from WT mouse or platelets from healthy individuals were isolated to measure PTPN22 expression by western blot with 2 different antibodies (A) or immunofluorescent staining (B). (C) PTPN22 expression in PTPN22−/− Plt was measured by western blot. Human megakaryocytic cell line (CMK) was used as human Mk. Data are representative of 3 independent experiments. Scar bar = 10 μm. (D) Tail bleeding time and FeCl3-induced arterial thrombosis in WT and PTPN22−/− mice (mean). Representative image of arterial thrombosis at different time point is shown. (E) Platelet adhesion on collagen under flow conditions. Whole blood was labeled with mepacrine and perfused through fibrillar collagen–coated BioFlux plates (Fluxion Biosciences) at 40 dynes/cm2 for 5 minutes. Platelet adhesion (covered area) was quantified at different time points (mean ± standard error, n = 6; one-way analysis of variance). **P < .01, ***P < .001, ****P < .0001.
Using PTPN22−/− mice, we further assessed the role of PTPN22 in platelet production. We found no significant differences in mean platelet volume, platelet distribution width, plateletcrit (supplemental Figure 3A), or expression of platelet receptors αIIbβ3, GPIbα, and GPVI (supplemental Figure 3B), nor in the number and size of α-granules and dense granules (supplemental Figure 3C), between WT mice and PTPN22−/− mice (P > .05). Consistently, PTPN22 deficiency did not affect in vitro megakaryocyte differentiation (supplemental Figure 4A), ploidy distribution (supplemental Figure 4B) or megakaryocyte differentiation into platelets (supplemental Figure 4C) under physiological conditions. Published studies have shown roles for numerous phosphatases in the regulation of megakaryocyte development.24-26 However, Holmes et al27 reported no differences in number or function of hematopoietic stem cell and progenitor populations in PTPN22-deficient mice under homeostatic conditions. Taken together with published data, our findings may indicate that PTPN22 activity is partially influenced by biorheological and vascular components present in the periphery.
Next, we performed a tail bleeding assay and FeCl3-induced mesenteric arterial thrombosis. We found significantly shortened tail bleeding time and enhanced arterial thrombus formation in PTPN22−/− mice compared WT mice (P < .01) (Figure 1D); these findings suggest that PTPN22 deficiency significantly promotes in vivo hemostasis and arterial thrombosis. To eliminate the contributing role of PTPN22 from other blood cells in arterial thrombus formation, we established a platelet depletion/reconstitution model and showed the consistent role of platelet PTPN22 in arterial thrombosis (supplemental Figure 5). Interestingly, the accelerated formation of arterial thrombus in PTPN22-deficient mice was significantly inhibited after administration of aspirin (supplemental Figure 6), suggesting that aspirin can inhibit the hyperreactivity of PTPN22-deficient platelets. In addition, we investigated the role of PTPN22 in venous thrombus formation using a deep vein thrombosis model. It is worth noting that PTPN22 deficiency did not affect venous thrombus formation, as shown by no differences in thrombus length (P > .05) or thrombus weight (P > .05) (supplemental Figure 7A). Consistently, PTPN22 deficiency did not affect the level of coagulation factors VIII and IX or the activated partial thromboplastin time (supplemental Figure 7B). Furthermore, under arterial flow conditions, PTPN22 deficiency significantly promoted in vitro thrombus formation (Figure 1E). From these data, it is indicated that PTPN22 deficiency promotes platelet hemostatic function and arterial thrombosis without affecting venous thrombosis.
PTPN22-deficient platelets exhibit enhanced platelet aggregation, granule secretion, αIIbβ3 activation, and calcium mobilization
PTPN22-deficient platelets presented significantly enhanced platelet aggregation compared with WT platelets after stimulation with CRP (0.1 μg/mL) (Figure 2A), collagen (0.25 μg/mL) (Figure 2B), thrombin (0.01 U/mL) (Figure 2C), and adenosine 5′-diphosphate (ADP) (5 μM) (Figure 2D) (P < .05). Consistently, the secretion of granules, including dense-granule (ATP release) (Figure 2A-C), α-granule (P-selectin expression) (Figure 2E), and ADP (Figure 2F), was significantly enhanced in PTPN22-deficient platelets after stimulation with CRP compared with those in WT platelets (P < .05). In addition, PTPN22-deficient platelets also displayed significantly enhanced integrin αIIbβ3 activation (higher JON/A binding) (Figure 2H) and calcium mobilization (Figure 2G) compared with WT platelets after stimulation. Using antibody arrays, we further detected the cytokines released from activated platelets and reported significantly higher levels of XCL1, macrophage colony-stimulating factor, P-selectin, stromal cell–derived factor 1α (SDF-1α), CCL-17, and CCL-1 released from PTPN22-deficient platelets than those released from WT platelets after stimulation (Figure 2I). Among those, SDF-1α and CCL-17 have been shown to activate platelets and amplify platelet activation.28,29 Therefore, the elevated levels of SDF-1α and CCL-17 may contribute to the hyperactivity of PTPN22-deficient platelets after agonist stimulation.
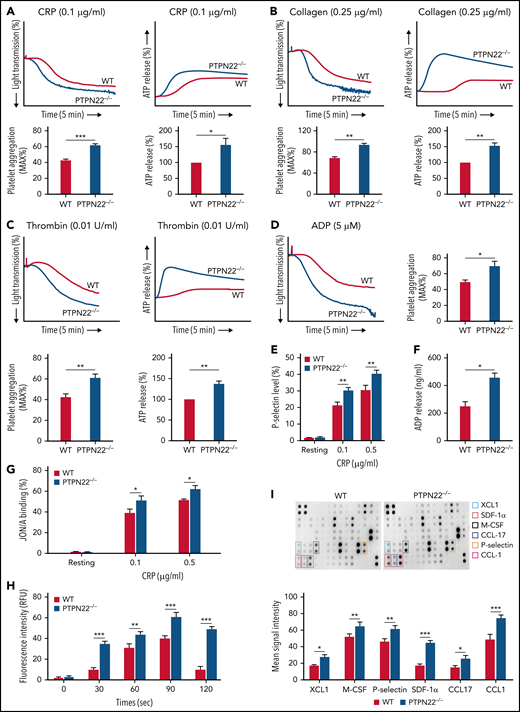
Platelet aggregation, granule recreation, and αIIbβ3 activation. Platelets (200 × 109/L) were isolated from WT or PTPN22−/− mice and stimulated with CRP (0.1 μg/mL) (A), collagen (0.25 μg/mL) (B), thrombin (0.01 U/mL) (C), or ADP (5 μM) (D) followed by analysis of platelet aggregation and ATP release (indicator of dense granule secretion) (except ADP stimulation) in a Model 700 Lumi-Aggregometer (Chrono-log Corporation). P-selectin expression (α-granule secretion) (presented as the % positive staining of anti-CD62P antibody) (E), ADP secretion (F), αIIbβ3 activation (presented by JON/A binding) (G), calcium mobilization (H), and cytokines released in response to CRP stimulation (I). Data are shown as mean ± standard error (n = 3-5; unpaired t test or two-way analysis of variance). *P < .05, **P < .01, ***P < .001. M-CSF, macrophage colony-stimulating factor; RFU, relative fluorescence units.
Platelet aggregation, granule recreation, and αIIbβ3 activation. Platelets (200 × 109/L) were isolated from WT or PTPN22−/− mice and stimulated with CRP (0.1 μg/mL) (A), collagen (0.25 μg/mL) (B), thrombin (0.01 U/mL) (C), or ADP (5 μM) (D) followed by analysis of platelet aggregation and ATP release (indicator of dense granule secretion) (except ADP stimulation) in a Model 700 Lumi-Aggregometer (Chrono-log Corporation). P-selectin expression (α-granule secretion) (presented as the % positive staining of anti-CD62P antibody) (E), ADP secretion (F), αIIbβ3 activation (presented by JON/A binding) (G), calcium mobilization (H), and cytokines released in response to CRP stimulation (I). Data are shown as mean ± standard error (n = 3-5; unpaired t test or two-way analysis of variance). *P < .05, **P < .01, ***P < .001. M-CSF, macrophage colony-stimulating factor; RFU, relative fluorescence units.
PTPN22 deficiency enhances platelet lamellipodia formation, spreading, and clot retraction
We then investigated whether PTPN22 affects αIIbβ3 outside-in signaling through measurement of platelet spreading and clot retraction, 2 processes regulated by early- and late-αIIbβ3 outside-in signaling, respectively.30 First, we evaluated the filopodia and lamellipodia formation of spread platelets by differential interference contrast microscopy and showed that PTPN22 deficiency significantly promoted lamellipodia formation (Figure 3A). Second, we analyzed the spread area by fluorescent microscopy and found that, compared with WT platelets, PTPN22-deficient platelets exhibited significantly increased platelet spreading on collagen or fibrinogen (Figure 3B). In accordance with this finding, thrombin-mediated clot retraction was also significantly enhanced in PTPN22-deficient platelets (Figure 3C).
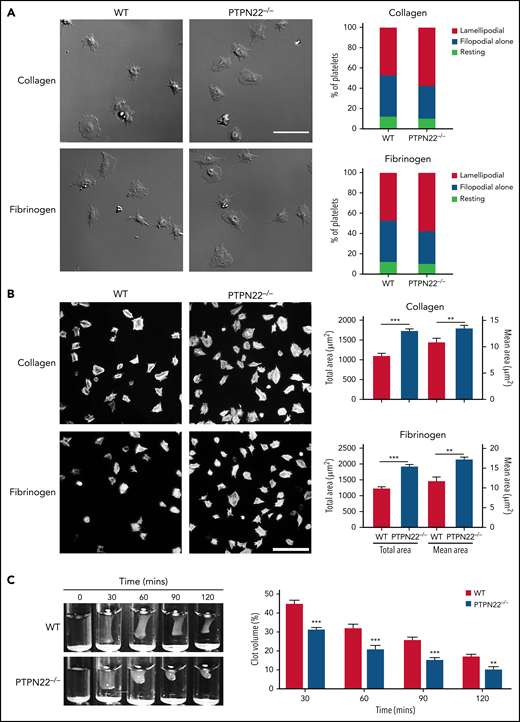
Platelet spreading and thrombin-mediated clot retraction. (A) Platelets (6 × 106/mL) were allowed to spread on collagen or fibrinogen for 20 minutes and fixed, followed by observation of filopodia and lamellipodia formation under a differential interference contrast microscope (Zeiss LSM 880) (×63 oil objective). (B) Platelets (12 × 106/mL) were placed on glass coverslips that were precoated with collagen or fibrinogen at 37°C for 90 minutes and then stained with Alexa Fluor-546–labeled phalloidin followed by analysis of the covered area using ImageJ software (National Institutes of Health). Images (×100) (scale bar = 20 μm) are representative of 3 independent experiments (mean ± standard deviation, n = 3; unpaired t test). (C) Clot retraction was triggered after addition of thrombin (1 U/mL). Representative images at 30, 60, 90, and 120 minutes from 3 independent experiments are presented. Data were quantified as clot volume (%) and are presented as mean (n = 3; two-way analysis of variance). **P < .01, ***P < .001.
Platelet spreading and thrombin-mediated clot retraction. (A) Platelets (6 × 106/mL) were allowed to spread on collagen or fibrinogen for 20 minutes and fixed, followed by observation of filopodia and lamellipodia formation under a differential interference contrast microscope (Zeiss LSM 880) (×63 oil objective). (B) Platelets (12 × 106/mL) were placed on glass coverslips that were precoated with collagen or fibrinogen at 37°C for 90 minutes and then stained with Alexa Fluor-546–labeled phalloidin followed by analysis of the covered area using ImageJ software (National Institutes of Health). Images (×100) (scale bar = 20 μm) are representative of 3 independent experiments (mean ± standard deviation, n = 3; unpaired t test). (C) Clot retraction was triggered after addition of thrombin (1 U/mL). Representative images at 30, 60, 90, and 120 minutes from 3 independent experiments are presented. Data were quantified as clot volume (%) and are presented as mean (n = 3; two-way analysis of variance). **P < .01, ***P < .001.
Protein phosphorylation profiles in PTPN22-deficient platelets after stimulation
To investigate the molecular mechanism by which PTPN22 deficiency alters platelet function, we analyzed protein phosphorylation profiles in platelets (Figure 4A) using quantitative phosphoproteomics. After CRP stimulation, 7119 phosphosites, 3654 phosphopeptides, and 1513 phosphoproteins (supplemental Data File 1) were identified, including some well-known kinases downstream of the GPVI signaling pathway, Syk, Lyn, and PLCγ2.

Quantitative phosphoproteomic analysis in WT and PTPN22−/− platelets after stimulation. (A) WT or PTPN22−/− platelets were treated with CRP for 1 minute followed by quantitative phosphoproteomic analysis. (B) Differentially expressed phosphopeptides between 2 groups as shown by volcano map. The x-axis indicates the fold change (logarithmic conversion based on 2) and y-axis indicates the P value (logarithmic conversion based on 10). Red dots presented the differentially expressed phosphopeptides with significance, and black dots indicate the phosphopeptides without significance. (C) The number of differentially expressed phosphopeptides with significance obtained from comparison of 2 groups. (D) Details of the differentially expressed phosphopeptides with significance identified from the comparison of the PC and the WC group. PC, PTPN22-deficient platelets after CRP stimulation; PH, pleckstrin homology; PR, PTPN22-deficient platelets under resting conditions; TNF, tumor necrosis factor; WC, wild-type platelets after CRP stimulation; WR, wild-type platelets under resting conditions.
Quantitative phosphoproteomic analysis in WT and PTPN22−/− platelets after stimulation. (A) WT or PTPN22−/− platelets were treated with CRP for 1 minute followed by quantitative phosphoproteomic analysis. (B) Differentially expressed phosphopeptides between 2 groups as shown by volcano map. The x-axis indicates the fold change (logarithmic conversion based on 2) and y-axis indicates the P value (logarithmic conversion based on 10). Red dots presented the differentially expressed phosphopeptides with significance, and black dots indicate the phosphopeptides without significance. (C) The number of differentially expressed phosphopeptides with significance obtained from comparison of 2 groups. (D) Details of the differentially expressed phosphopeptides with significance identified from the comparison of the PC and the WC group. PC, PTPN22-deficient platelets after CRP stimulation; PH, pleckstrin homology; PR, PTPN22-deficient platelets under resting conditions; TNF, tumor necrosis factor; WC, wild-type platelets after CRP stimulation; WR, wild-type platelets under resting conditions.
Using a threshold (a fold change >1.2 or <0.83 and P < .05), we identified the differentially expressed phosphopeptides in WT and PTPN22-deficient platelets (supplemental Data File 2). A total of 204 upregulated and 199 downregulated phosphopeptides were found in WT platelets and 200 upregulated and 192 downregulated phosphopeptides in PTPN2-deficient platelets after stimulation (Figure 4B). Meanwhile, comparison of WT and PTPN22−/− platelets revealed that 6 upregulated and 32 downregulated phosphopeptides were observed under resting conditions relative to 9 upregulated and 18 downregulated phosphopeptides after stimulation. In addition, Kyoto Encyclopedia of Genes and Genomes pathway analysis revealed that phosphoproteins related to these differentially expressed phosphopeptides were enriched in platelet activation, tumor necrosis factor signaling pathway, FcεRI signaling pathway, MAPK signaling pathway, and Ras signaling pathway (supplemental Figure 8).
To verify a potential target protein to be part of a regulated signaling cascade and thus meaningful, candidate proteins should respond to signals from agonists and therefore have increased phosphorylation upon stimulation in the PTPN22−/− platelets. However, as dynamic regulation of protein phosphorylation within platelets after stimulation, at a given time point, the phosphorylation status of certain proteins is likely to be differentially maintained or dephosphorylated due to negative-feedback systems. It would therefore be more valuable and informative to temporally evaluate the phosphorylation status of proteins in the quantitative phosphoproteomic assay to obtain the dynamic profile of changes to protein phosphorylation. In our phosphoproteomic assay, platelets were treated with CRP for 1 minute followed by the proteomic analysis. Thus, to avoid the loss of some key proteins identified in the proteomic assay, we analyzed all the dysregulated phosphorylated proteins (4 + 7+1 + 0+4 + 5) between WT and PTPN22-deficient platelets after stimulation rather than only proteins with increased stimulation-dependent phosphorylation that were increased in PTPN22−/− platelets (4 + 7+1 + 0) (Figure 4C) alone. After analysis, there were 7 upregulated phosphopeptides with differential expression following comparison of PTPN22-deficient platelets with WT platelets after CRP stimulation (Figure 4D). These were P39428 (tumor necrosis factor receptor–associated factor 1, TRAF1, Ser283, Tyr290, Thr291); O70469 (docking protein 2, Dok2, Tyr53); Q63918 (caveolae-associated protein 2, Cavin2, Ser287); Q8CG03 (guanosine 3′,5′-cyclic monophosphate [cGMP]-specific 3′,5′-cyclic phosphodiesterase, PDE5A, Ser92); P70302 (stromal interaction molecule 1, STIM1, Ser521); Q9JM52 (misshapen-like kinase 1, Mink1, Ser740); and P52734 (FYVE, RhoGEF and PH domain-containing protein 1, Fgd1, Ser98). Among these phosphopeptides, PDE5A,31 STIM1,32 and MINK122 have been shown to play roles in platelet function. However, the phosphorylation sites within STIM1 (Ser521) and MINK1 (Ser740) have not previously been reported or characterized, and their roles in the regulation of platelet function are unknown. In addition, STIM1 deficiency does not affect platelet aggregation or thrombus formation in vitro at arterial or venous shear rates.33 However, the phosphorylation of PDE5A (Ser92) has been shown to be associated with increased catalytic degradation of cGMP.34,35 Considering the fundamental role of PDE5A phosphorylation at Ser92 in the regulation of platelet function through selective hydrolysis of cGMP, we elected to focus on PDE5A in subsequent experiments.
Increased PDE5A phosphorylation and reduced cGMP signaling in PTPN22-deficient platelets after stimulation
As only a single time point sample was analyzed in our proteomic analysis, to obtain the full profile of the changes of PDE5A phosphorylation, we performed western blotting analysis of PDE5A phosphorylation (Ser92) at different time points after CRP or thrombin stimulation. Before this, the specificity of the phospho-PDE5A (Ser92) antibody was evaluated by using Phospho-PDE5A blocking peptide, and the results showed that p-PDE5A was not recognized and detected by Phospho-PDE5A (Ser92) antibody after blocking peptide treatment, confirming the specificity of this antibody (supplemental Figure 9). As seen in Figure 5A, PDE5A phosphorylation reached a peak at 15 seconds after CRP stimulation, followed by a decrease in the following time points without difference at 1 minute compared with resting WT platelets. Similarly, PTPN22-deficient platelets also displayed the same trend of PDE5A phosphorylation with no difference between resting and 1 minute. This is why the dysregulated PDE5A phosphorylation was not identified in the quantitative phosphoproteomic assay between wild-type platelets after CRP stimulation vs wild-type platelets under resting condition as well as PTPN22-deficient platelets after CRP stimulation vs PTPN22-deficient platelets under resting condition (Figure 4C). However, a significant difference in PDE5A phosphorylation between PC vs WC (PTPN22-deficient platelets vs WT platelets after stimulation) was consistently identified by the proteomic assay and western blot. Similarly, significantly increased PDE5A phosphorylation (Ser92) was found in PTPN22-deficient platelets after stimulation with thrombin (Figure 5B) compared with WT platelets (P < .01), further confirming enhanced PDE5A phosphorylation in PTPN22-deficient platelets.
![Increased PDE5A phosphorylation in PTPN22-deficient platelets. WT or PTPN22−/− platelets were treated with CRP (5 μg/mL) (A) for 5, 15, 30, or 60 seconds or thrombin (1 U/mL) (B) for 1, 3, or 5 minutes followed by measurement of PDE5A phosphorylation (Ser92) and expression by western blot (mean ± standard deviation [SD], n = 4 independent experiments; two-way analysis of variance [ANOVA]). (C) WT or PTPN22−/− platelets were isolated and stimulated with CRP (5 μg/mL) for 1 minute and then the level of cGMP and cyclic adenosine monophosphate (cAMP) was measured by using an enzyme-linked immunosorbent assay kit (mean ± standard error [SE], n = 5-7; t test). (D) After stimulation with CRP (5 μg/mL), the phosphorylation of vasodilator-stimulated phosphoprotein (VASP) (Ser157 and Ser239) was measured (mean ± SD, n = 4 independent experiments; two-way ANOVA). PTPN22−/− platelets were pretreated with vehicle or 10 μM sildenafil (PDE5A inhibitor) for 1 hour at 37°C followed by measurement of platelet aggregation in response to CRP (0.25 μg/mL) (mean ± SE, n = 4; one-way ANOVA) (E), P-selectin expression in response to CRP (0.5 μg/mL) (mean ± SE, n = 3; one-way ANOVA) (F), or spreading on collagen (mean ± SD, n = 3; one-way ANOVA) (scale bar = 10 μm) (G). *P < .05, **P < .01, ***P < .001. ns, no significance.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/9/10.1182_blood.2022015554/3/m_bloodbld2022015554f5.png?Expires=1765003338&Signature=c3EgOds~oMHXGYeEa0bVFb8XT8-moOOJdlw8KL65sVCsH9YFlKX3sajhqBaCUl~1lGYQ-I8MUMPbG82icyHuJTYzpdZjaID5-FCOR38rxewuu~Rn2cHp~rGARhG1Nn8qaX0VaGHpaLZBdT0l4MD~Li~Vq6HUnWjQwEbeibZ4Z4jVGmMaHjyRqkWUudk9Rl5QqCZgf-aXBn8uVWvFEExqpwltb1CrhQ-A14-BClCS5SVQAivN~UMWKNTtafz10uvc5sKR5Y82afJftufcjEpVIVhfNzQbcG-FiLJqYJnsqB-7t6WZ0u3ol78b7fPnp1t62rJ8j4V6yFVQsg715iKeyQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Increased PDE5A phosphorylation in PTPN22-deficient platelets. WT or PTPN22−/− platelets were treated with CRP (5 μg/mL) (A) for 5, 15, 30, or 60 seconds or thrombin (1 U/mL) (B) for 1, 3, or 5 minutes followed by measurement of PDE5A phosphorylation (Ser92) and expression by western blot (mean ± standard deviation [SD], n = 4 independent experiments; two-way analysis of variance [ANOVA]). (C) WT or PTPN22−/− platelets were isolated and stimulated with CRP (5 μg/mL) for 1 minute and then the level of cGMP and cyclic adenosine monophosphate (cAMP) was measured by using an enzyme-linked immunosorbent assay kit (mean ± standard error [SE], n = 5-7; t test). (D) After stimulation with CRP (5 μg/mL), the phosphorylation of vasodilator-stimulated phosphoprotein (VASP) (Ser157 and Ser239) was measured (mean ± SD, n = 4 independent experiments; two-way ANOVA). PTPN22−/− platelets were pretreated with vehicle or 10 μM sildenafil (PDE5A inhibitor) for 1 hour at 37°C followed by measurement of platelet aggregation in response to CRP (0.25 μg/mL) (mean ± SE, n = 4; one-way ANOVA) (E), P-selectin expression in response to CRP (0.5 μg/mL) (mean ± SE, n = 3; one-way ANOVA) (F), or spreading on collagen (mean ± SD, n = 3; one-way ANOVA) (scale bar = 10 μm) (G). *P < .05, **P < .01, ***P < .001. ns, no significance.
Increased PDE5A phosphorylation in PTPN22-deficient platelets. WT or PTPN22−/− platelets were treated with CRP (5 μg/mL) (A) for 5, 15, 30, or 60 seconds or thrombin (1 U/mL) (B) for 1, 3, or 5 minutes followed by measurement of PDE5A phosphorylation (Ser92) and expression by western blot (mean ± standard deviation [SD], n = 4 independent experiments; two-way analysis of variance [ANOVA]). (C) WT or PTPN22−/− platelets were isolated and stimulated with CRP (5 μg/mL) for 1 minute and then the level of cGMP and cyclic adenosine monophosphate (cAMP) was measured by using an enzyme-linked immunosorbent assay kit (mean ± standard error [SE], n = 5-7; t test). (D) After stimulation with CRP (5 μg/mL), the phosphorylation of vasodilator-stimulated phosphoprotein (VASP) (Ser157 and Ser239) was measured (mean ± SD, n = 4 independent experiments; two-way ANOVA). PTPN22−/− platelets were pretreated with vehicle or 10 μM sildenafil (PDE5A inhibitor) for 1 hour at 37°C followed by measurement of platelet aggregation in response to CRP (0.25 μg/mL) (mean ± SE, n = 4; one-way ANOVA) (E), P-selectin expression in response to CRP (0.5 μg/mL) (mean ± SE, n = 3; one-way ANOVA) (F), or spreading on collagen (mean ± SD, n = 3; one-way ANOVA) (scale bar = 10 μm) (G). *P < .05, **P < .01, ***P < .001. ns, no significance.
To selectively evaluate agonist-induced PDE5A phosphorylation, platelets were treated with an αIIbβ3 inhibitor (tirofiban) to block integrin outside-in signaling. We found that inhibition of αIIbβ3 did not affect PDE5A phosphorylation in either CRP- or thrombin-stimulated platelets (supplemental Figure 10), indicating that PDE5A phosphorylation is independent of integrin αIIbβ3 activation. Because PDE5A phosphorylation plays a role in the regulation of nucleotide levels via hydrolysis of cyclic adenosine monophosphate and cGMP,31 we then analyzed their level in activated platelets and found significantly lower cGMP levels in PTPN22-deficient platelets than in WT platelets after CRP stimulation (P < .01) (Figure 5C). Interestingly, there was no difference in cyclic adenosine monophosphate levels between WT and PTPN22-deficient platelets after CRP treatment (P < .05); this is consistent with the role of PDE5A phosphorylation in the selective hydrolysis of cGMP,36 indicating an impaired cGMP signaling pathway in PTPN22-deficient platelets. Consistently, the phosphorylation of vasodilator-stimulated phosphoprotein (Ser157/239), which is a substrate of cGMP-dependent protein kinase,37 was significantly decreased in PTPN22-deficient platelets after CRP treatment compared with WT platelets (P < .05) (Figure 5D). However, other signaling pathways such as protein kinase C, extracellular signal-regulated kinase 1/2, and AKT were not affected by PTPN22 deficiency (P > .05) (supplemental Figure 11). To further investigate whether the increased PDE5A phosphorylation contributes to the enhanced function of PTPN22-deficient platelets, WT or PTPN22−/− platelets were treated with sildenafil, a PDE5A inhibitor, and significantly decreased platelet aggregation (Figure 5E), degranulation (Figure 5F), and spreading on collagen (Figure 5G) were observed after treatment. The treatment induced no difference in WT platelets, suggesting that PTPN22 negatively regulates platelet function, possibly via regulation of PDE5A.
PTPN22 interacts with p-PDE5A and dephosphorylates it in activated platelets
Because enhanced PDE5A phosphorylation was observed in PTPN22-deficient platelets after stimulation, a coimmunoprecipitation assay was performed to further investigate whether PTPN22 interacts with PDE5A in platelets after activation. It showed that PTPN22 binds p-PDE5A in CRP-stimulated platelets (Figure 6A). Because PTPN22 has been shown to interact with Csk in T cells,38 a kinase involved in platelet function though inhibition of SFKs,39 we also investigated whether PTPN22 interacts with Csk in platelets. Surprisingly, PTPN22 did not interact with Csk in either resting or activated platelets. To further verify the interaction between PTPN22 and p-PDE5A, the reverse coimmunoprecipitation was conducted to determine if anti–p-PDE5A antibody could pull-down PTPN22. We observed that p-PDE5A could also bind PTPN22 in activated platelets (Figure 6B), further confirming the interaction between PTPN22 and p-PDE5A.

Interaction of PTPN22 with p-PDE5A in activated platelets. Coimmunoprecipitation analysis of the relationship of PTPN22 with p-PDE5A or Csk (A) or the relationship of p-PDE5A with PTPN22 (B) in platelets after stimulation with CRP (5 μg/mL) for 5, 15, 30, or 60 seconds. (Similar results were obtained from 3 independent experiments). (C) Lysates of CRP-treated PTPN22-deficient platelets were incubated with GST-PTPN22 PTPase (0.4 μg) (in the presence or absence of 40 μM LTV-1), GST-PTPN22 C227S, or GST followed by measurement of PDE5A phosphorylation (Ser92) by western blot. —: indicates the PDE5A phosphorylation in CRP-treated platelets. (D) After CRP stimulation, PTPN22-deficient platelet lysates were incubated with GST or GST-PTPN22 PTPase followed by detection of the phosphorylation of PDE5A (Ser92) or Syk (Tyr519/520) by western blot. The phosphorylation of PDE5A or Syk after GST-PTPN22 PTPase treatment was quantified as a ratio of their phosphorylation level relative to GST (mean ± SD, n = 3 independent experiments). **P < .01. Phosphopeptide (1 μM) was incubated with increasing amounts of GST or GST-PTPN22 PTPase (0-16 ng) (E), or GST and GST-PTPN22 PTPase (4 ng) was incubated with the increasing amounts of peptide (0-4 μM) for 5 minutes at 37°C followed by the addition of 50 μL Malachite Green Reagent and measurement of the absorption value (mean ± standard error, n = 3 independent experiments) (F). (G) GST-PTPN22 PTPase (4 ng) was incubated with peptide (1 μM) in the absence (vehicle) or presence of LTV-1 (20 μM) or NC1 (20 μM) for 5 minutes followed by measurement of phosphate release. The phosphate concentration was calculated by using the phosphate standard and corrected by subtracting the values of GST protein. Mutant: PTPN22 C227S. Compared with vehicle, ***P < .001 (mean ± standard error, n = 3 independent experiments).
Interaction of PTPN22 with p-PDE5A in activated platelets. Coimmunoprecipitation analysis of the relationship of PTPN22 with p-PDE5A or Csk (A) or the relationship of p-PDE5A with PTPN22 (B) in platelets after stimulation with CRP (5 μg/mL) for 5, 15, 30, or 60 seconds. (Similar results were obtained from 3 independent experiments). (C) Lysates of CRP-treated PTPN22-deficient platelets were incubated with GST-PTPN22 PTPase (0.4 μg) (in the presence or absence of 40 μM LTV-1), GST-PTPN22 C227S, or GST followed by measurement of PDE5A phosphorylation (Ser92) by western blot. —: indicates the PDE5A phosphorylation in CRP-treated platelets. (D) After CRP stimulation, PTPN22-deficient platelet lysates were incubated with GST or GST-PTPN22 PTPase followed by detection of the phosphorylation of PDE5A (Ser92) or Syk (Tyr519/520) by western blot. The phosphorylation of PDE5A or Syk after GST-PTPN22 PTPase treatment was quantified as a ratio of their phosphorylation level relative to GST (mean ± SD, n = 3 independent experiments). **P < .01. Phosphopeptide (1 μM) was incubated with increasing amounts of GST or GST-PTPN22 PTPase (0-16 ng) (E), or GST and GST-PTPN22 PTPase (4 ng) was incubated with the increasing amounts of peptide (0-4 μM) for 5 minutes at 37°C followed by the addition of 50 μL Malachite Green Reagent and measurement of the absorption value (mean ± standard error, n = 3 independent experiments) (F). (G) GST-PTPN22 PTPase (4 ng) was incubated with peptide (1 μM) in the absence (vehicle) or presence of LTV-1 (20 μM) or NC1 (20 μM) for 5 minutes followed by measurement of phosphate release. The phosphate concentration was calculated by using the phosphate standard and corrected by subtracting the values of GST protein. Mutant: PTPN22 C227S. Compared with vehicle, ***P < .001 (mean ± standard error, n = 3 independent experiments).
Considering that PTPN22 is a tyrosine phosphatase, we investigated whether there are any tyrosine phosphorylation sites on PDE5A through immunoprecipitation of PDE5A and blotting with phosphotyrosine antibody. The results revealed no tyrosine phosphorylation of PDE5A in CRP-treated platelets, as shown by no protein bands around 100 kDa (ie, PDE5A molecular weight) (supplemental Figure 12), indicating that there are no tyrosine phosphorylation sites on PDE5A; this is consistent with a previous study showing no other phosphorylation site besides Ser92.40 Consistently, a substrate trapping experiment using trapping mutant of PTPN22 (D195A/C227S, DACS)10 found no interaction between the trapping mutant and PDE5A in platelets in the absence or presence of pervanadate, as indicated by no positive protein bands around 100 kDa (the molecular weight of PDE5A) after blotting the membrane with phosphotyrosine antibody (supplemental Figure 13).
To further investigate the catalytic activity of PTPN22 on PDE5A, we performed an in vitro protein phosphatase assay through incubation of lysates of CRP-treated PTPN22−/− platelets with purified GST-PTPN22 PTPase, the catalytically inactive mutant form of PTPN22 (GST-PTPN22 C227S), or GST. We found dephosphorylation of p-PDE5A after treatment with GST-PTPN22 PTPase (Figure 6C) but not after GST or the mutant form of treatment.
Because PTPN22 is a tyrosine phosphatase, we also evaluated whether PTPN22 possesses phosphatase activity on tyrosine phosphorylation, and the results showed the decrease of Syk phosphorylation (Tyr519/520) in CRP-stimulated platelets after treatment with GST-PTPN22 PTPase (Figure 6D). Interestingly, PTPN22 seems to exert a relatively higher phosphatase activity on serine phosphorylation over tyrosine phosphorylation in platelets. To further verify this, we performed another in vitro phosphatase activity assay using phosphopeptide. The results indicated that PTPN22 could induce the dephosphorylation of both pSer (p-PDE5A) and pTyr (p-Syk) peptides with a relatively higher phosphatase activity on serine phosphorylation over tyrosine phosphorylation, as shown by the increased release of phosphate measured by using the Malachite Green assay (Figure 6E-F). However, the dephosphorylation of pSer or pTyr peptide induced by PTPN22 was completely inhibited by the addition of PTPN22 inhibitor LTV-1 or NC1 (Figure 6G).
PTPN22 inhibition enhances human platelet function
To investigate whether PTPN22 also plays a role in human platelet function, human platelets were treated with LTV-1, a PTPN22 inhibitor. Prior to this, we evaluated the specificity of LTV-1 using PTPN22-deficient platelets and showed that addition of LTV-1 did not affect platelet aggregation and clot retraction in PTPN22-deficient platelets (supplemental Figure 14). Inhibition of PTPN22 by LTV-1 significantly increased platelet aggregation in response to CRP (0.1 μg/mL) or thrombin (0.02 U/mL) stimulation compared with vehicle treatment (Figure 7A). Meanwhile, platelet spreading on collagen or fibrinogen (Figure 7B) as well as the lamellipodia formation (supplemental Figure 15) were also significantly enhanced in LTV-1–treated platelets along with accelerated thrombin-mediated clot retraction (Figure 7C), consistent with the negatively regulatory role of PTPN22 in mouse platelet function. Moreover, we also found significantly elevated PDE5A phosphorylation levels after LTV-1 treatment compared with vehicle treatment (P < .01) (Figure 7D), indicating that PTPN22 might also negatively regulate human platelet function through regulation of PDE5A phosphorylation.
![The role of PTPN22 role in human platelet function. Washed human platelets were isolated and pretreated with 20 μM LTV-1 (PTPN22 inhibitor) for 1 hour at 37°C followed by analysis of platelet aggregation after stimulation with CRP (0.1 μg/mL) or thrombin (0.02 U/mL) (mean ± standard deviation [SD], n = 3; t test) (A), platelet spreading on collagen or fibrinogen (mean ± SD, n = 3; t test) (scale bar = 10 μm) (B), and clot retraction induced by thrombin (0.8 U/mL) (mean ± SD, n = 3; two-way analysis of variance) (C). (D) PDE5A phosphorylation was also measured in platelets stimulated with CRP 5 μg/mL in the presence or absence of LTV-1 (mean ± SD, n = 3 independent experiments; two-way analysis of variance). **P < .01, ***P < .001.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/9/10.1182_blood.2022015554/3/m_bloodbld2022015554f7.png?Expires=1765003338&Signature=VSwe5iSqWEskObdvG1qTkFRAl5d3critjtT~PPJyUu7s66NvMbhALwR99Pwp97C95lTgXE5tbxBmmdPvFE67MLuV5oJIBVKrL-CJgQyHb9i3yKHkw2~Ih5Fh5STIF7PwMlu6evBQa2C-AGcamJlZ73iscLebPL5IPM3WnrbPbTteGuRH2CxdxF3uc4nfaNa0X9kv1HW7P3TuBwj-lTBhVa4wENE43RvnTPcY~cmufUui8K2FGi-QUIvslXi0lbKiToWMk5tZtR53QuAXRMAtB77hKHpe2dtko4o7wiEBzckkEs8Lmm1iYRIXtGhswPssPIhpEcVHS~YHa~~RjCoikQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
The role of PTPN22 role in human platelet function. Washed human platelets were isolated and pretreated with 20 μM LTV-1 (PTPN22 inhibitor) for 1 hour at 37°C followed by analysis of platelet aggregation after stimulation with CRP (0.1 μg/mL) or thrombin (0.02 U/mL) (mean ± standard deviation [SD], n = 3; t test) (A), platelet spreading on collagen or fibrinogen (mean ± SD, n = 3; t test) (scale bar = 10 μm) (B), and clot retraction induced by thrombin (0.8 U/mL) (mean ± SD, n = 3; two-way analysis of variance) (C). (D) PDE5A phosphorylation was also measured in platelets stimulated with CRP 5 μg/mL in the presence or absence of LTV-1 (mean ± SD, n = 3 independent experiments; two-way analysis of variance). **P < .01, ***P < .001.
The role of PTPN22 role in human platelet function. Washed human platelets were isolated and pretreated with 20 μM LTV-1 (PTPN22 inhibitor) for 1 hour at 37°C followed by analysis of platelet aggregation after stimulation with CRP (0.1 μg/mL) or thrombin (0.02 U/mL) (mean ± standard deviation [SD], n = 3; t test) (A), platelet spreading on collagen or fibrinogen (mean ± SD, n = 3; t test) (scale bar = 10 μm) (B), and clot retraction induced by thrombin (0.8 U/mL) (mean ± SD, n = 3; two-way analysis of variance) (C). (D) PDE5A phosphorylation was also measured in platelets stimulated with CRP 5 μg/mL in the presence or absence of LTV-1 (mean ± SD, n = 3 independent experiments; two-way analysis of variance). **P < .01, ***P < .001.
Discussion
PTPN22 is a PTP, and it inhibits T-cell function through dephosphorylating key kinases of the T cell–signaling pathway.38 Previous studies identified PTPN22 transcript in megakaryocyte using transcriptome-based approaches.14,15 In our study, we first demonstrated expression of PTPN22 in both human and mouse platelets. Subsequent functional studies using PTPN22−/− mice showed that PTPN22 plays a negative role in platelet function and thrombosis.
Currently, several phosphatases have been identified in platelets, such as CD148, PTP-1B, and Shp1/2. Using a genetic mouse model, CD148 has been shown to be a master positive regulator of SFKs through direct dephosphorylation of the activation loop in platelets, and its deficiency caused impaired platelet aggregation and spreading, in vivo bleeding, and reduced thrombus formation.41,42 Apart from its role in metabolic disorders or cancer,43,44 PTP-1B positively regulates platelet integrin αIIbβ3 outside-in signaling through displacing Csk from a complex formed by b3 and Src, leading to subsequent activation of Src.45 The exact role of Shp1/2 in the regulation of platelet function is much more complex, as they might play distinct roles in platelet function even though they are structurally related. Shp1 positively regulates SFKs downstream of GPVI and aIIbb324 but negatively regulates G protein–coupled receptor signaling in platelets,46 whereas Shp2 inhibits GPVI-dependent signaling transduction47 and negatively regulates platelet responses mediated by CLEC-2 and αIIbβ324 as well as thrombus stability under high shear stress.48 In this study, we described the enhanced function of PTPN2-deficient platelets.
Quantitative phosphoproteomics analysis found 3654 phosphopeptides and 1513 phosphoproteins, including Syk, Lyn, and PLCγ2 (well-known kinases downstream of GPVI signaling). These findings are consistent with a previous study showing >3000 significant phosphorylation events on >1300 proteins over conditions initiating and progressing GPVI-mediated platelet activation in human platelets after CRP stimulation by phosphoproteomic quantitation.49 Through comparison of the difference between WT and PTPN22-deficient platelets, we then identified 5 differentially expressed phosphopeptides and selected PDE5A as the potential target of PTPN22. In vitro validation assay revealed that PTPN22 deficiency significantly enhanced PDE5A phosphorylation in CRP-stimulated platelets. Meanwhile, we also observed the reduction of cGMP level and vasodilator-stimulated phosphoprotein phosphorylation in PTPN22-deficient platelets, possibly due to the increase in PDE5A phosphorylation, which has been shown to be capable of selectively hydrolyzing cGMP to serve as important feedback to limit the amplitude and duration of cGMP signaling in platelets.36
Because alteration of PDE5A phosphorylation was found in PTPN22-deficient platelets after stimulation, we speculate that PDE5A might be a target of PTPN22. To verify this hypothesis, we first performed the coimmunoprecipitation assay and found that PTPN22 binds p-PDE5A in activated platelets. The subsequent in vitro dephosphorylation assay using the lysates of CRP-stimulated PTPN22−/− platelets or phosphopeptide with PDE5A Ser92 motif showed that PTPN22 could induce PDE5A dephosphorylation at Ser92. In addition, PTPN22 also dephosphorylates Syk phosphorylation at Tyr519/520 in CRP-stimulated platelets or phosphopeptide of Syk with Tyr519/520 phosphorylation. Interestingly, in neutrophils, PTPN22 deficiency does not affect Syk phosphorylation at Tyr519/520 but significantly impairs the Fcγ receptor–mediated neutrophil activation,50 which is opposite its role in platelets or lymphocytes as a negative regulator, possibly due to the different substrates of PTPN22 in different cell types.
In conclusion, our study shows that PTPN22 is expressed in platelets and negatively regulates platelet function and arterial thrombus formation, identifying new potential targets for future prevention of thrombotic defects.
Acknowledgments
The authors thank Steve P. Watson (University of Birmingham) for critically reading the manuscript and providing valuable comments. They also acknowledge Shanghai Applied Protein Technology Co., Ltd. for technological assistance in the quantitative phosphoproteomic analysis.
This work was supported by the National Natural Science Foundation of China (grant numbers 82170130, 81970124, 81400082, 81641151, and 81700178), the Natural Science Foundation of Jiangsu Province (grant numbers BK20140219 and BK20170259), funding for the Distinguished Professorship Program of Jiangsu Province, the Shuangchuang Project of Jiangsu Province, the Six Talent Peaks Project of Jiangsu Province (WSN-133), the “333 Project” of Jiangsu Province (BRA2017542), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (18KJA320010 and 17KJA320008), Jiangsu Province’s Key Provincial Talents Program (ZDRCA2016054), Jiangsu Province’s Graduate Scientific Research Innovation Program (KYCX18-2186, KYCX19-2231, and KYCX19-2234), the Youth Science and Technology Innovation Team of Xuzhou Medical University, and the National Health and Medical Research Council, Australia.
Authorship
Contribution: X.W., G.W., and Y.D. performed research, analyzed data, and wrote the manuscript; X.G., H.T., X.X., Sixuan Zhang, Z.S., W.J., Y.L., R.Y., Q.W., Z. Lu, C.F., Z. Li, and Si Zhang performed experiments and analyzed data; E.E.G. and R.K.A. provided the intellectual input and wrote the manuscript; H.H. analyzed data and wrote the manuscript; and L.Z., K.X., and J.Q. conceived and designed the study and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jianlin Qiao, Blood Diseases Institute, Xuzhou Medical University, 84 West Huaihai Rd, Xuzhou 221002, China; e-mail: jianlin.qiao@gmail.com.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal