

In this issue of Blood, Wang et al1 describe that in platelets an obscure protein, tyrosine phosphatase non-receptor type 22 (PTPN22), acts as a negative regulator of signaling transduction.
This study improves our understanding of the function of tyrosine phosphatases in platelets, which potentially also applies to other blood cells. In early days, platelet signaling was considered an easy business, although this was not realized. It was thought that protein tyrosine kinases mediated platelet activation, and protein tyrosine phosphatases were doing the opposite. Later, with the discovery that the receptor-type tyrosine phosphatase PTPRJ (CD148) enhances rather than decreases glycoprotein VI (GPVI)- and collagen-induced platelet activation via Src-family kinases, complexity increased.2 The paper from Wang et al provides a new chapter to our understanding of how protein tyrosine phosphatases (PTPs) can work in platelets.
In general terms, PTPs are known as signaling proteins that regulate a multitude of cellular processes, including cell growth, differentiation, mitosis, metabolism, signaling, and oncogenic transformation.3 To date, the genome-wide analysis of the platelet and megakaryocyte transcriptome and proteome has revealed relevant expression levels of as many as 54 tyrosine protein phosphatases.4 These include 20 isoforms of the dual specificity phosphatases (DUSP1-DUSP28), and 18 isoforms of the receptor-type tyrosine protein phosphatases (PTPRA-PTPRU). In addition, 16 PTPN isoforms (PTPN1-PTPN21) have been identified, which can be divided into high, medium, or low expression (see figure). Often, but not always, PTPN isoforms cause a negative regulation of specific tyrosine kinase-induced signaling reactions, for instance in insulin or T-cell signaling.3 In other words, absence of the PTPN (in mouse) or a loss-of-function mutation of the PTPN (in man) often is accompanied by increased tyrosine kinase-mediated signal transduction.
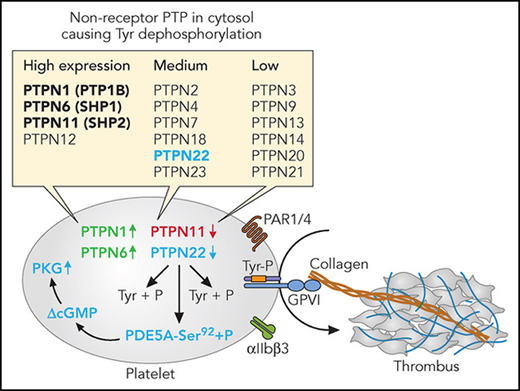
Non-receptor protein tyrosine phosphates (PTPN) and platelet activation. Indicated are PTPN isoforms with high, medium, or low expression in both human and mouse platelets (bold: previously studied). In red, negative regulator of thrombus formation; in green, positive regulator; in blue, negative regulation pathway proposed by Wang et al. Professional illustration by Patrick Lane, ScEYEnce Studios.
Non-receptor protein tyrosine phosphates (PTPN) and platelet activation. Indicated are PTPN isoforms with high, medium, or low expression in both human and mouse platelets (bold: previously studied). In red, negative regulator of thrombus formation; in green, positive regulator; in blue, negative regulation pathway proposed by Wang et al. Professional illustration by Patrick Lane, ScEYEnce Studios.
For platelets, this has been investigated in reasonable detail for three of the four most abundantly expressed isoforms, PTPN1, PTPN6, and PTPN11 (see figure). The results of those studies were surprising, in that genetic deletion of Ptpn1 or Ptptn6 (the latter to a larger extent) led to a reduction in platelet responses.5,6 Under flow conditions, absence of PTPN1 caused reduced collagen/GPVI-dependent thrombus formation and integrin αIIbβ3 signaling. In contrast, megakaryocyte deficiency in mouse Ptpn11 resulted in macrothrombocytopenia and platelet hyper-responsiveness,7 such in agreement with evidence for a negative role of the human protein in platelet activation.8 Furthermore, patients with the Noonan syndrome and a gain-of-function mutation in PTPN11 experience platelet dysfunction, reduced thrombus formation, and a bleeding phenotype.8
In the new paper by Wang et al, the authors have used a clever approach to unravel a mechanism behind the negative role on platelets of mouse Ptpn22, which is a medium-level expressed PTPN isoform in both mouse and human platelets. In Ptpn22-deficient mice, they found an increased platelet response to low doses of collagen, thrombin, and ADP, accompanied by increased platelet spreading. Analysis of the phosphoproteome of the knockout mouse platelets, activated via GPVI, showed seven differentially regulated phosphopeptides (based on statistically confirmed cutoff levels of 20%), of which the most interesting peptide corresponded to Ser92 of guanosine 3′,5′-cyclic monophosphate (cGMP)-specific phosphodiesterase 5A (PDE5A). In extensive follow-up experiments, they found that the P-Ser92 site is a direct target of PTPN22, showing lower phosphorylation than in wild-type mouse platelets, which effect was accompanied by an increased cGMP level (see figure). Indeed, elevated cGMP is a key mechanism for platelet inhibition via protein kinase G (PKG).9 Why is this work novel and interesting? For three reasons: the surprisingly large phenotype given the medium expression of PTPN22 in mouse platelets; the new identification of a PTPN22 substrate (ieIe, PDE5A), explaining its negative role in platelet activation irrespective of agonist type; and the fact that the PTPN22 substrate sequence contains a phospho-serine rather than a phospho-tyrosine.
Is this the end of a PTPN22-platelet story? Likely not, as there remain several open questions. First, a primary endothelial-related phenotype in the Ptpn22 knockout, affecting platelet cGMP levels, cannot be completely ruled out. Second, because mass spectrometric phosphoproteomic analysis with stable isotope labeling requires a multistep work-up, data precision hence is an issue. Hence, other phospho-tyrosine targets explaining the platelet phenotype might have been missed. Third, in both the mouse and human platelet phosphoproteomes, the region of Ser78-Thr96 of PDE5A can be (ant)agonist-regulated. Yet, this region is located far from the active site binding cGMP, which in mouse PDE5A is located at residue Ala807. Finally, unlike the isoform PDE3A, PDE5A is not considered as the key regulator of (human) platelet function, which agrees with the limitedeffect on platelets of the widely used drug, sildenafil.10 Though in favor of the authors’ scheme, the PTPN22 inhibitor LTV-1 enhanced the aggregation of human platelets.1 Logical follow-up animal experiments would be the generation of a megakaryocyte-specific knockout or of a CRISPR mouse containing a mutated PDE5A phospho-site. Stay tuned!
Conflict-of-interest disclosure: J.W.M.H. is connected to the Synapse Research Institute Maastricht.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal