

In this issue of Blood, Vats et al1 report a mechanism of lung injury in sickle cell disease (SCD) initiated by gasdermin D–driven production of neutrophil extracellular traps (NETs) in the liver that travel to, and occlude, the pulmonary microcirculation.
Obstruction of the microvasculature is a major feature of SCD and is characterized by a multistep process involving adhesive intercellular interactions of blood cells within the activated endothelium.2 Acute vasoocclusive pain episodes are the leading cause of hospital admissions for SCD and an important determinant of disease severity.3 These episodes often precede a severe type of acute lung injury, termed acute chest syndrome, which represents the main cause of mortality among individuals with SCD.4
Pulmonary complications are associated with occlusions of the pulmonary vasculature caused by aggregates of neutrophils, platelets, and erythrocytes. In vivo studies have shown that P-selectin, an adhesion molecule expressed by activated platelets and endothelial cells, activates immune cells and drives intercellular adhesion and SCD vasoocclusion.5,6 These findings led to the development of a humanized P-selectin antibody approved by the Food and Drug Administration (FDA) for use in patients with SCD. A clinical trial reported a 45% reduction in the frequency of vasoocclusive events among patients with SCD receiving this anti–P-selectin therapy.7 Consistently, blocking P-selectin conferred protection from pulmonary vasoocclusion in mice with SCD8 but did not completely abrogate the occurrence of such events. Beyond a favorable outcome, these findings suggest that P-selectin–mediated adhesion is not the sole mechanism triggering systemic and pulmonary vasoocclusion in SCD.
In the present study, Vats et al elegantly uncover a P-selectin–independent mechanism underlying vasoocclusion within the lung vasculature, which could explain the uncomplete efficacy of anti–P-selectin therapies. Hemolysis derivatives, such as cell-free oxygenated hemoglobin (oxy-Hb), are potent damage-associated molecular patterns (DAMPs) that promote endothelial dysfunction and sterile inflammation and can precipitate vasoocclusion.9 Using an oxy-Hb–induced vasoocclusion protocol in a SCD mouse model, the authors identify large neutrophil-platelet aggregates occluding the pulmonary arterioles and marked abundance of NETs in the lungs, when compared with controls (ie, mice with SCD administered saline or control mice administered oxy-Hb).
Although the participation of NETs in SCD lung vasoocclusion has been reported,10 a striking finding from the Vats study is that NETs found in the lungs were not produced locally but originated elsewhere and traveled through the circulation. Using state-of-the-art live imaging of the pulmonary microcirculation, the authors visualized NETs arriving through the pulmonary arterioles and, notably, this was independent of the canonical P-selectin signaling because the number of circulating NETs was not different in the lungs of mice lacking this receptor. In contrast to the lungs, imaging the liver microcirculation of mice with SCD revealed a large number of neutrophils releasing NETs, which was even higher when mice were challenged with oxy-Hb. By interrupting the blood flow from the liver to the lungs through clamping of the hepatic artery and the portal vein, the authors found drastic reductions in the number of circulating NETs reaching the lungs. Although this strategy does not exclude other sources of NETs upstream from the liver, the impressive set of images and elegant strategies used by the authors strongly support their hypothesis that NETs promoting pulmonary vasoocclusion are generated remotely and travel intravascularly to the lungs. The data also provide striking demonstration of a type of embolism that involves transfer of a substrate for secondary immunothrombotic activation (the NETs) and vasoocclusion (see figure), which we speculate can be frequent in other forms of vascular inflammation.
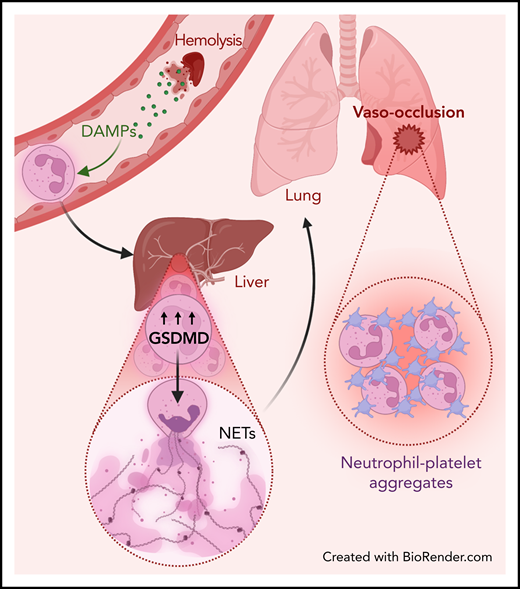
Liver-to-lung NET embolization in SCD. Hemolysis releases DAMPs that prime neutrophils and lead to the activation of an interferon and gasdermin-D (GSDMD)-dependent signaling pathway, which promotes NET generation into the liver microcirculation. These NETs carried through the blood into the lungs can then promote neutrophil-platelet aggregation, leading to pulmonary vasoocclusion.
Liver-to-lung NET embolization in SCD. Hemolysis releases DAMPs that prime neutrophils and lead to the activation of an interferon and gasdermin-D (GSDMD)-dependent signaling pathway, which promotes NET generation into the liver microcirculation. These NETs carried through the blood into the lungs can then promote neutrophil-platelet aggregation, leading to pulmonary vasoocclusion.
Mechanistic insights into this NET-induced vasoocclusion in lungs came from the identification of upregulated type I interferon signaling in neutrophils from mice with SCD. Downstream genes were upregulated as well, including caspase-11, the ortholog to human caspase-4, and gasdermin-D, an effector of inflammasome signaling and therefore a valuable indicator of sterile inflammation. Both were undetectable in control mouse neutrophils but were found in neutrophils from human patients with SCD, which upregulated caspase-4 and gasdermin-D, thereby providing proof of remarkable interspecies conservation of this signaling axis. A series of in vitro experiments using both human and mouse neutrophils further demonstrated that activation of this pathway was triggered by oxygen reactive species, which are known to be abundant upon hemolysis. In vivo, the authors found that inhibition of gasdermin-D reduced liver to lung embolization of circulating NETs and prevented SCD lung vasoocclusion, which, again, was shown to be independent on P-selectin.
These findings not only expand our understanding of the vasoocclusive pathophysiology but also may inspire new therapeutic approaches for the prevention of these episodes and secondary complications in patients with SCD. Although hematologists currently count on several FDA-approved drugs for treating SCD, namely hydroxyurea, l-glutamine, crizanlizumab, and voxelotor, none of them appear to manage the broad spectrum of complications, to overcome the variable response among patients, or to prevent end-organ damage when used as monotherapies. It is likely that managing the complexity of SCD pathophysiology will demand a multidrug approach that allows combined targeting of the different instigating pathways as demonstrated in this study. Indeed, Vats et al show that when both mechanisms are targeted by inhibiting gasdermin-D in mice lacking P-selectin after oxy-Hb treatment, occlusion of the pulmonary microcirculation is completely abolished. Thus, it will be urgent to explore whether these exciting findings translate into therapies to improve the lives of thousands of patients afflicted with this debilitating disease. Likewise, it will be important to examine whether similar mechanisms of intravascular immune activation and thromboinflammatory dissemination, driven by DAMPs, are involved in the myriad of pathologies that undermine the health of our circulatory system.
Conflict-of-interest disclosure: A.H. is a paid consultant for Flashipt Pioneering, Inc for matters unrelated to this paper. L.S.T. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal