


Abstract
High-grade B-cell lymphoma (HGBL), not otherwise specified (NOS), is a recently introduced diagnostic category for aggressive B-cell lymphomas. It includes tumors with Burkitt-like or blastoid morphology that do not have double-hit cytogenetics and that cannot be classified as other well-defined lymphoma subtypes. HBCLs, NOS, are rare and heterogeneous; most have germinal center B-cell phenotype, and up to 45% carry a single-hit MYC rearrangement, but otherwise, they have no unifying immunophenotypic or cytogenetic characteristics. Recent analyses using gene expression profiling (GEP) revealed that up to 15% of tumors currently classified as diffuse large B-cell lymphoma display an HGBL-like GEP signature, indicating a potential to significantly expand the HGBL category using more objective molecular criteria. Optimal treatment of HGBL, NOS, is poorly defined because of its rarity and inconsistent diagnostic patterns. A minority of patients have early-stage disease, which can be managed with standard R-CHOP–based approaches with or without radiation therapy. For advanced-stage HGBL, NOS, which often presents with aggressive disseminated disease, high lactate dehydrogenase, and involvement of extranodal organs (including the central nervous system [CNS]), intensified Burkitt lymphoma–like regimens with CNS prophylaxis may be appropriate. However, many patients diagnosed at age >60 years are not eligible for intensive immunochemotherapy. An improved GEP- and/or genomic-based pathologic classification that could facilitate HGBL-specific trials is needed to improve outcomes for all patients. In this review, we discuss the current clinicopathologic concept of HGBL, NOS, and existing data on its prognosis and treatment and delineate potential future taxonomy enrichments based on emerging molecular diagnostics.
Introduction
The classification of aggressive B-cell lymphomas has evolved, driven by the delineation of subtypes with unique clinical, pathologic, and molecular features. Accurate classification is important to clinicians, who translate it into refinement of therapeutic approaches. For decades, pathologists have recognized that some B-cell neoplasms morphologically resemble Burkitt lymphoma (BL), while displaying phenotypic or genotypic characteristics inconsistent with this diagnosis. Clinically, such cases behave more aggressively than diffuse large B-cell lymphoma (DLBCL), which led to their provisional designation as high-grade B-cell lymphoma (HGBL), Burkitt-like, in the 1996 Revised European-American Lymphoma Classification.1 The differentiation of HGBL from BL or DLBCL remained problematic, with disagreement between experts in half of cases.2
In 2008, the World Health Organization (WHO) reclassified these high-grade tumors as “B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and BL”3 (BCLU). The BCLU description still emphasized morphology, despite discovery of Burkitt-like gene expression profiles (GEPs) and MYC rearrangements (MYC-Rs).4-6 BCLU further included cases with blastoid morphology, a gray zone between lymphoblastic lymphoma and DLBCL. Interestingly, pediatric hematologists have tailored chemotherapy in aggressive B-cell lymphomas to the clinical presentation (eg, stage, lactate dehydrogenase [LDH] level, and bone marrow or central nervous system [CNS] involvement) rather than specific histology, whereas adult oncologists have strictly separated BL and DLBCL, variably pooling HGBL with either group.7
Confronted with the poor prognosis of lymphomas carrying concurrent MYC-R and BCL2 (and/or BCL6) rearrangement, the 2016 WHO classification separated these double (DHLs)/triple-hit lymphomas as a formal HGBL category.8-11 Consequently, the BCLU subtype was broken apart, and non-DHL tumors were designated as HGBL, not otherwise specified (NOS).12 This entity is again defined by morphologic criteria. The WHO advocated that HGBL, NOS, be diagnosed sparingly and “only when the pathologist is truly unable to confidently classify the case as DLBCL or BL.”12(p340) The resulting diagnostic variability hinders translational and clinical research, with a paucity of prognostic or treatment data for these highly aggressive cancers.
In this review, we discuss HGBL, NOS, in the context of emerging molecular and clinical insights. We examine how the definition of HGBL could be reformulated using cytogenetic and molecular criteria, as was first done with DHL, with the goal of establishing specific therapeutic approaches. Hematologists are challenged by treatment decisions for aggressive, rapidly progressing lymphomas, and trials that have used intensified therapy beyond R-CHOP (rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone) for clinically high-risk DLBCL have been unsuccessful in improving survival.13-17 Recognition of molecular HGBL may offer a chance to better identify the most aggressive B-cell lymphomas and to delineate optimal therapy through dedicated clinical trials.
HGBL, NOS, in the WHO classification
According to the 2016 WHO classification, HGBL, NOS, includes B-cell tumors with either BL-like or blastoid morphology that do not meet criteria for other well-defined categories (Figure 1).12 Adequate classification requires evaluation by cytomorphology, immunohistochemistry, and fluorescence in situ hybridization (FISH). DHL should be classified as HGBL with concurrent MYC-R and BCL2 and/or BCL6 rearrangement. Morphologically clear-cut DLBCL with a single-hit MYC-R should be classified as DLBCL, NOS, regardless of the proliferative fraction or other molecular and cytogenetic features.12,18,19 Historically worse prognosis in DLBCL with MYC-R was largely driven by DHL,9,10,20 and the Lunenburg Lymphoma Biomarker Consortium study of 2383 DLBCLs showed no significant difference in outcomes between DLBCL with or without a single-hit MYC-R.21 Extra copies of MYC have been variably associated with worse prognosis in DLBCL/BCLU.22-24 Extra copies of MYC, coexpression of MYC and BCL2 by immunohistochemistry (dual-expressor lymphoma [DEL]), and alterations in TP53 or other genes are not currently considered in the WHO definition of HGBL, NOS.
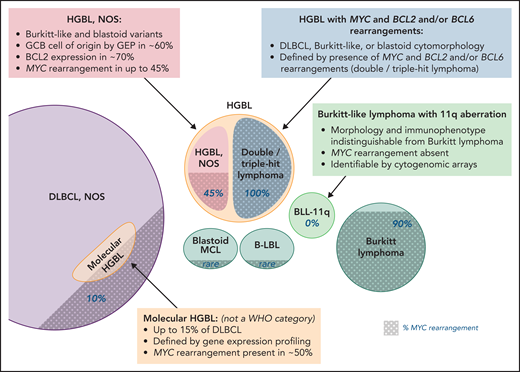
HGBL, NOS and other aggressive B-cell lymphomas. Note that the size of the circles is not to scale, as the precise incidence of some entities is unknown; shaded areas indicate the proportion of cases with MYC rearrangement. Professional illustration by Patrick Lane, ScEYEnce Studios.
HGBL, NOS and other aggressive B-cell lymphomas. Note that the size of the circles is not to scale, as the precise incidence of some entities is unknown; shaded areas indicate the proportion of cases with MYC rearrangement. Professional illustration by Patrick Lane, ScEYEnce Studios.
In a pathologic study of 32 HGBL, NOS, cases from the German High Grade Lymphoma Study Group, CD10 was expressed in 65%, BCL6 in 91%, and BCL2 in 65%, and MYC-R was present in 13%.25MYC-R may be accompanied by BCL2 amplifications.18,26 An inverse pattern with MYC amplification and BCL2-IGH translocation, or amplification of both oncogenes, can also be encountered.27 Among 50 HGBL, NOS, cases from the University of Pittsburgh (Burkitt-like, n = 41; blastoid, n = 9), 8% showed MYC-R, 32% MYC extra copies, and 60% no MYC alterations.28 In a recent Chinese series of 41 cases, 76% expressed CD10, 83% BCL6, 63% MUM1/IRF4, and 71% MYC.29
The Lymphoma/Leukemia Molecular Profiling Project study provided the first molecular characterization of HGBL, NOS.30 Notably, 53% of 64 centrally reviewed cases were reclassified as DLBCL or BL, confirming the poor reproducibility of the WHO criteria. There were no significant differences in cytogenetic, GEP, or genomic features between confirmed HGBL, NOS, cases and cases reclassified as DLBCL. Among confirmed HGBLs, 57% had germinal center B-cell (GCB) origin by GEP, but 25% were activated B-cell tumors. MYC-R was present in 46%, BCL2 in 10%, and BCL6 in 12%; the most common mutations involved KMT2D (43%) and TP53 (30%). Mutation-based subtypes and GEP signatures were variable, suggesting that HGBL, NOS, contains several biologic entities, including some activated B-cell tumors with MYD88, CD79B, or TBL1XR1 mutations.
Burkitt-like variant
The Burkitt-like variant of HGBL, NOS, corresponds directly to the historical category of BCLU. It is a GCB (CD10+ BCL6+) lymphoma characterized by Burkitt-like morphology (diffuse infiltrate of intermediate-sized cells, tingible body macrophages, and generally high proliferation rate; Figure 2) but often more pleomorphic appearance and sometimes lower Ki-67 staining (50% to 90%). Other cases are morphologically indistinguishable from BL, yet may show expression of BCL2, lack CD10 expression, or have multiple cytogenetic abnormalities or other characteristics unusual for BL. Of note, 5% to 10% of true BLs may not show MYC-R, so tumors with otherwise classical morphology and immunophenotype should be designated as BL.12,31
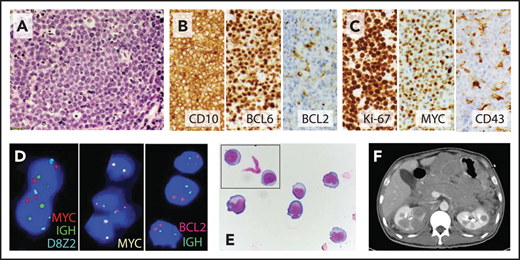
Burkitt-like variant of HGBL, NOS. (A) High magnification of hematoxylin and eosin stain shows a starry-sky pattern with a diffuse infiltrate of medium to large cells with finely dispersed chromatin, several small nucleoli, and frequent mitoses. (B) Malignant B cells express the germinal center markers CD10 and BCL6 but are largely negative for BCL2. (C) Ki-67 proliferation index was ∼100%, and neoplastic cells showed high expression of MYC but were CD43−; other negative markers included BCL1, TdT, MUM1, and EBER by in situ hybridization. (D) FISH shows no evidence of MYC-R or BCL2-IGH rearrangement (FISH probes: Vysis LSI IGH/MYC/CEP 8 Tri-Color Dual Fusion Probe, Vysis LSI MYC-Dual Color Break-Apart Probe, and Vysis LSI IGH/BCL2 Dual Color Dual Fusion Probe; Abbott Molecular). (E) Cerebrospinal fluid cytology shows medium to large cells with round to irregular nuclear contours, variably prominent nucleoli, and moderate amount of cytoplasm. (F) The patient, a 43-year-old HIV− man, presented with extensive abdominal adenopathy and infiltration of the CNS, gastrointestinal tract, bilateral kidneys, and adrenal glands on computed tomography scan; he attained a complete response (CR) to the R-CODOX-M/IVAC (rituximab plus cyclophosphamide, doxorubicin, vincristine, and high-dose methotrexate alternating with ifosfamide, cytarabine, and etoposide) regimen with intrathecal therapy and remains in remission 1 year later. Original magnification: ×600 (A), ×400 (B-C), and ×1000 (E).
Burkitt-like variant of HGBL, NOS. (A) High magnification of hematoxylin and eosin stain shows a starry-sky pattern with a diffuse infiltrate of medium to large cells with finely dispersed chromatin, several small nucleoli, and frequent mitoses. (B) Malignant B cells express the germinal center markers CD10 and BCL6 but are largely negative for BCL2. (C) Ki-67 proliferation index was ∼100%, and neoplastic cells showed high expression of MYC but were CD43−; other negative markers included BCL1, TdT, MUM1, and EBER by in situ hybridization. (D) FISH shows no evidence of MYC-R or BCL2-IGH rearrangement (FISH probes: Vysis LSI IGH/MYC/CEP 8 Tri-Color Dual Fusion Probe, Vysis LSI MYC-Dual Color Break-Apart Probe, and Vysis LSI IGH/BCL2 Dual Color Dual Fusion Probe; Abbott Molecular). (E) Cerebrospinal fluid cytology shows medium to large cells with round to irregular nuclear contours, variably prominent nucleoli, and moderate amount of cytoplasm. (F) The patient, a 43-year-old HIV− man, presented with extensive abdominal adenopathy and infiltration of the CNS, gastrointestinal tract, bilateral kidneys, and adrenal glands on computed tomography scan; he attained a complete response (CR) to the R-CODOX-M/IVAC (rituximab plus cyclophosphamide, doxorubicin, vincristine, and high-dose methotrexate alternating with ifosfamide, cytarabine, and etoposide) regimen with intrathecal therapy and remains in remission 1 year later. Original magnification: ×600 (A), ×400 (B-C), and ×1000 (E).
Blastoid variant
The blastoid variant of HGBL, NOS, is rare. It has intermediate-sized cells that show blastoid morphology (medium nuclei with inconspicuous nucleoli and finely dispersed chromatin) while retaining the mature (TdT− CD20+) immunophenotype (Figure 3). Blastoid HGBL, NOS, must be distinguished from B-lymphoblastic lymphoma (expressing TdT or CD34) and blastoid mantle cell lymphoma (defined by expression of cyclin D1, SOX11, and/or CCND1-IGH rearrangement). Otherwise, this entity remains poorly characterized. CD10, BCL6, and BCL2 are frequently, but not unvaryingly, expressed, whereas MYC or BCL2 rearrangement is uncommon (excluding DHL).28,32 Some cases represent transformation from follicular lymphoma or other indolent B-cell lymphomas.33 In the largest study of 24 cases (including blastoid DHL), 85% had GCB phenotype, 30% lacked CD10, 14% expressed CD5, and 3 of 4 tested cases stained for IRF4/MUM1.32 Furthermore, 89% had complex karyotype.
![Blastoid variant of HGBL, NOS. (A) Hematoxylin and eosin stain shows an infiltrate of medium blastoid cells with finely dispersed chromatin and inconspicuous nucleoli; touch preparation (inset) showed neoplastic cells with mostly rounded nuclei, inconspicuous nucleoli, and scant cytoplasm; the cells expressed CD19, CD20, PAX5, CD10, BCL6, and Ki-67 (>95%), but not MYC (<10%), cyclin D1, TdT, or BCL2. Additionally, stains from MUM1/IRF4, CD15, CD30, CD23, and PD-L1 were negative. (B) FISH showed normal pattern for MYC, BCL2, and IGH, but 3 copies of BCL6 (Vysis LSI IGH/BCL2 Dual Color Dual Fusion Probe, Vysis LSI BCL6 [ABR] Dual Color Break Apart Rearrangement Probe, and Vysis LSI MYC-Dual Color Break-Apart Probe; Abbott Molecular). (C) The patient, a 33-year-old woman, presented with a large mediastinal mass histologically and immunophenotypically inconsistent with B-lymphoblastic or primary mediastinal B-cell lymphoma; she attained a CR with 6 cycles of DA-EPOCH-R (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) and remains in remission 3 years after therapy. Original magnification: ×600 (A).](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/140/9/10.1182_blood.2020008374/3/m_bloodbld2020008374cf3.png?Expires=1770058732&Signature=KbwckwzQoh22tTD4pdqaAUNwlCtb6Egkqct7w2QwJSIbhZ8sAFfJ7Ibv54nQAnX8~SX8B28yNZSLBNvgp6SI5F91KFLgcgUtKL3w-KSI20bpBb6-o2EacQ4UFg6pfWnpKGdv5g33JMItxnhviBPvZdGM79MU4sL8AU4mIsGI3meFicDdEclMgmprlWrRcrFHXNpLBlHodu0xQPYbsYkwtncOJelYi00WZRIEbPFaiEdezQAgTGKXnPN4VIUXNGZT27r6bYESUAI3tLs9cGB-8JQIrP7SrwI1XNdSOZ0esm4kD6Bn2mVgmIwHlP7dPn3ENG0c5F4LlhnRIbLXkcemFg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Blastoid variant of HGBL, NOS. (A) Hematoxylin and eosin stain shows an infiltrate of medium blastoid cells with finely dispersed chromatin and inconspicuous nucleoli; touch preparation (inset) showed neoplastic cells with mostly rounded nuclei, inconspicuous nucleoli, and scant cytoplasm; the cells expressed CD19, CD20, PAX5, CD10, BCL6, and Ki-67 (>95%), but not MYC (<10%), cyclin D1, TdT, or BCL2. Additionally, stains from MUM1/IRF4, CD15, CD30, CD23, and PD-L1 were negative. (B) FISH showed normal pattern for MYC, BCL2, and IGH, but 3 copies of BCL6 (Vysis LSI IGH/BCL2 Dual Color Dual Fusion Probe, Vysis LSI BCL6 [ABR] Dual Color Break Apart Rearrangement Probe, and Vysis LSI MYC-Dual Color Break-Apart Probe; Abbott Molecular). (C) The patient, a 33-year-old woman, presented with a large mediastinal mass histologically and immunophenotypically inconsistent with B-lymphoblastic or primary mediastinal B-cell lymphoma; she attained a CR with 6 cycles of DA-EPOCH-R (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) and remains in remission 3 years after therapy. Original magnification: ×600 (A).
Blastoid variant of HGBL, NOS. (A) Hematoxylin and eosin stain shows an infiltrate of medium blastoid cells with finely dispersed chromatin and inconspicuous nucleoli; touch preparation (inset) showed neoplastic cells with mostly rounded nuclei, inconspicuous nucleoli, and scant cytoplasm; the cells expressed CD19, CD20, PAX5, CD10, BCL6, and Ki-67 (>95%), but not MYC (<10%), cyclin D1, TdT, or BCL2. Additionally, stains from MUM1/IRF4, CD15, CD30, CD23, and PD-L1 were negative. (B) FISH showed normal pattern for MYC, BCL2, and IGH, but 3 copies of BCL6 (Vysis LSI IGH/BCL2 Dual Color Dual Fusion Probe, Vysis LSI BCL6 [ABR] Dual Color Break Apart Rearrangement Probe, and Vysis LSI MYC-Dual Color Break-Apart Probe; Abbott Molecular). (C) The patient, a 33-year-old woman, presented with a large mediastinal mass histologically and immunophenotypically inconsistent with B-lymphoblastic or primary mediastinal B-cell lymphoma; she attained a CR with 6 cycles of DA-EPOCH-R (etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin, and rituximab) and remains in remission 3 years after therapy. Original magnification: ×600 (A).
Apart from BL and B-lymphoblastic and blastoid mantle cell lymphomas, another entity to be differentiated from HGBL, NOS, is Burkitt-like lymphoma with 11q aberration (BLL-11q), a provisional WHO subtype morphologically indistinguishable from BL.34-36 BLL-11q is predominantly nodal, lacks MYC-R, and occurs mostly in children or younger adults or as a posttransplantation lymphoproliferative disorder.36,37 It is difficult to identify in practice, because its defining cytogenetic lesion (amplification of 11q23.2-q23.3 with telomeric deletion of 11q24.1-qter) is missed by standard karyotyping. It may be suggested by presence of extra copies of ATM (11q22.3) or KMT2A (11q23.3), but cytogenomic arrays or specialized FISH probes are necessary for confirmation. Approximately 5% to 20% of HGBLs, NOS, can be reclassified as BLL-11q.38 11q abnormalities are also found in straightforward BL or other HGBL.39 BLL-11q notably lacks the distinctive BL mutations in ID3, TCF3, MYC, CCND3, or TP53, so it may be more related to HGBL than to BL.34,35,40
Expanding concept: molecular HGBL
Recent analyses using GEP have advanced our understanding of HGBL, revealing that some tumors currently diagnosed as DLBCL have striking biologic similarities to HGBL. Emerging data confirm that these molecular high-grade (MHG) lymphomas have worse prognosis with R-CHOP therapy and might benefit from a refined classification. In 2006, GEP showed that 10% of DLBCLs and most BCLUs express a molecular BL signature characterized by overexpression of MYC targets, with decreased expression of major histocompatibility complex (MHC) genes and NF-κB signaling.4,5 Molecular BL carried more copy number alterations, 17p deletions, and MYC amplifications than classic BL.41,42 Nevertheless, most DLBCLs with a single-hit MYC-R were not molecular BL.
In 2019, this concept was expanded, uncovering that 10% to 25% of DLBCL tumors exhibit distinct GEP signatures similar to high-grade lymphomas (BL or DHL).43,44 Sha et al43 applied a GEP classifier to 928 DLBCLs from the phase 3 REMoDL-B trial (R-CHOP ± bortezomib), identifying 9% with the MHG signature (Table 1). MHG lymphomas were of GCB origin and 49% carried MYC-Rs, but only 36% were DHLs. MHG lymphomas showed low expression of MHC-II genes and downregulation of immune response pathways and were enriched in mutations in MYC, BCL2, DDX3X, TP53, and KMT2D.43,45 Three-year progression-free survival (PFS) was only 37% after R-CHOP, significantly worse than for either GCB or non-GCB DLBCL. MHG lymphomas carried poor prognosis regardless of MYC-R or DHL status. Their unfavorable outcomes after R-CHOP (5-year overall survival [OS], 44%) was confirmed in a UK population-based study.46
Characteristics of molecular HGBL in studies using GEP for patients diagnosed as having DLBCL
| Characteristic | (REMoDL-B)43 N = 928 | (BCCA)44 N = 157 (GCB only) |
|---|---|---|
| Study cohort | Phase 3 clinical trial | Observational cohort |
| Molecular HGBL, n (%) | 83 (9) | 42 (27) |
| Median age, y | 65 | 62 |
| Stage III/IV, n (%) | 63 (76) | 23 (56) |
| LDH | Median, 561 units/L | 58% elevated |
| Extranodal involvement, n (%) | 53 (64) | NR; 7% >1 site |
| High IPI, n (%) | 25 (30) | 7 (17) |
| GCB by GEP, % | 90 | 100 (by definition) |
| Cytogenetics, n (%) MYC-R Double/triple hit | 35 (49) 26 (36) | 27 (64) 22 (52) |
| Immunohistochemistry, n (%) | ||
| MYC+ BCL2+ MYC+/BCL2+ | 20 (71) 22 (79) 17 (61) | 30 (75) 36 (88) 25 (63) |
| PFS or TTP, % | 37 at 3 y (R-CHOP) vs 78 in GCB DLBCL vs 64 in ABC DLBCL | 57 at 5 y vs 81 in GCB DLBCL vs 51 in ABC DLBCL |
| OS, % | 57 at 3 y (all patients) vs 88 in GCB DLBCL vs 81 in ABC DLBCL | 60 at 5 y vs 81 in GCB DLBCL vs 56 in ABC DLBCL |
| GEP signature | Trained on BL vs DLBCL ↑ MYC, TCF3 targets ↓ MHC-II ↓ immune response ↓ inflammation | Trained on DHL vs DLBCL ↑ MYC, E2F targets ↓ MHC-I and MHC-II ↑ oxidative phosphorylation ↓ TNFα, NF-κB, IL-6/JAK/STAT3 |
| Mutations | MYC, BCL2, TP53, EZH2, DDX3X, KMT2D | MYC, BCL2, TP53, EZH2, DDX3X, KMT2D, CREBBP |
| Characteristic | (REMoDL-B)43 N = 928 | (BCCA)44 N = 157 (GCB only) |
|---|---|---|
| Study cohort | Phase 3 clinical trial | Observational cohort |
| Molecular HGBL, n (%) | 83 (9) | 42 (27) |
| Median age, y | 65 | 62 |
| Stage III/IV, n (%) | 63 (76) | 23 (56) |
| LDH | Median, 561 units/L | 58% elevated |
| Extranodal involvement, n (%) | 53 (64) | NR; 7% >1 site |
| High IPI, n (%) | 25 (30) | 7 (17) |
| GCB by GEP, % | 90 | 100 (by definition) |
| Cytogenetics, n (%) MYC-R Double/triple hit | 35 (49) 26 (36) | 27 (64) 22 (52) |
| Immunohistochemistry, n (%) | ||
| MYC+ BCL2+ MYC+/BCL2+ | 20 (71) 22 (79) 17 (61) | 30 (75) 36 (88) 25 (63) |
| PFS or TTP, % | 37 at 3 y (R-CHOP) vs 78 in GCB DLBCL vs 64 in ABC DLBCL | 57 at 5 y vs 81 in GCB DLBCL vs 51 in ABC DLBCL |
| OS, % | 57 at 3 y (all patients) vs 88 in GCB DLBCL vs 81 in ABC DLBCL | 60 at 5 y vs 81 in GCB DLBCL vs 56 in ABC DLBCL |
| GEP signature | Trained on BL vs DLBCL ↑ MYC, TCF3 targets ↓ MHC-II ↓ immune response ↓ inflammation | Trained on DHL vs DLBCL ↑ MYC, E2F targets ↓ MHC-I and MHC-II ↑ oxidative phosphorylation ↓ TNFα, NF-κB, IL-6/JAK/STAT3 |
| Mutations | MYC, BCL2, TP53, EZH2, DDX3X, KMT2D | MYC, BCL2, TP53, EZH2, DDX3X, KMT2D, CREBBP |
ABC, activated B cell; BCCA, British Columbia Cancer Agency; IL-6, interleukin-6; IPI, International prognostic index; NR, not reported; TNFα, tumor necrosis factor α; TTP, time to progression.
Ennishi et al44 studied 157 GCB DLBCL tumors, identifying 42 (27%) with a DHL-like GEP signature, although 50% were not cytogenetically DHLs (Table 1). These tumors universally expressed CD10, 64% had MYC-R, and 63% were DELs. The DHL-like lymphomas showed fewer tumor-infiltrating T cells, lower expression of MHC-I genes, and frequent point mutations in MYC, BCL2, DDX3X, TP53, and KMT2D. Importantly, compared with other GCB DLBCLs, 5-year PFS after R-CHOP was worse in DHL-like lymphomas regardless of cytogenetic DHL status. Whole-exome sequencing of non-DHL lymphomas with DHL-like GEP showed that 30% carried cryptic MYC and BCL2 translocations.47 In the Lymphoma/Leukemia Molecular Profiling Project study, the DHL-like GEP signature was present in 54% of HGBL, NOS, cases and in 39% of tumors reclassified as DLBCL.30
These seminal discoveries suggest that a well-defined subgroup of GCB tumors currently classified as DLBCL are in fact closer to HGBL from both molecular and clinical perspectives. Furthermore, molecular HGBL includes both DHL and non-DHL cases that are prognostically equivalent. If formally recognized in future taxonomy, the molecularly defined HGBL population would become large enough for subtype-specific clinical trials. As described, it requires GEP for identification, which is not available in current practice and would be challenging to implement for rapidly progressing tumors. However, recent sequencing data suggest a potential for genomic-based classification. Wright et al48 reported that the signatures described by Sha et al43 and Ennishi et al44 largely correspond to a subset of genomic EZB tumors enriched in MYC, TP53, DDX3X, GNA13, and FOXO1 mutations. The Haematological Malignancy Research Network study confirmed that subdividing the EZB/C3 cluster according to presence of MYC mutations identifies GCB tumors (EZB-MYC) with a particularly poor prognosis.49MYC mutations with MYC overexpression also defined the highest-risk GCB subtype in the study by Reddy et al.50
Other genetically defined forms of HGBL have been proposed and might further expand the molecular HGBL category. One subtype includes tumors with concurrent MYC-R and TP53 alteration (deletion or mutation).26,51TP53 alterations can serve as an alternative second hit to produce high-grade disease with complex karyotype and dismal outcomes.51-53 In 1 series, HGBL, NOS, morphology and poor survival were associated with MYC-R in combination with either TP53 mutation or p53 overexpression.53 In another analysis, the high-grade GEP signature carried poor prognosis only with concurrent TP53 abnormalities.54 A second category includes aggressive activated B-cell DLBCLs with MYD88, CD79B, and TBL1XR1 mutations and CDKN2A/B deletions (subset of MCD/C5 tumors) characterized by immune evasion and propensity for extranodal and CNS involvement.48,55-57 Approximately 9% of HGBLs, NOS, are identified as MCD by the LymphGen classifier, but further research is needed to characterize this subgroup.30
Therapy and outcomes
Clinical characteristics and outcomes
Clinicopathologic series have described HGBL, NOS, as a disease of mostly older adults (median age, ∼70 years), equally frequent in men and women, and often with adverse risk factors (high LDH, high IPI, extranodal involvement, and CNS invasion). In the published BCLU/HGBL series (all <45 cases, often including DHL), 55% to 68% of patients had stage III/IV disease, 60% to 100% high LDH, and 40% to 55% IPI ≥3, and one-third >1 involved extranodal site.29,58-62 Reported 2-year PFS rates ranged from 23% to 69%, and OS rates from 30% to 77%. There are no reliable data on prognostic factors specific to HGBL, NOS, although prognosis appears better with low IPI (0-2).29,58,62
First-line therapy
So far, prospective clinical trials have pooled HGBL, NOS, together with other aggressive B-cell lymphomas. Therefore, clinical practice relies on extrapolations and subset analyses (Table 2). Recently, 2 single-arm studies (using DA-EPOCH-R and R-CHOP plus lenalidomide) have specifically enrolled patients with MYC-R tumors, including some with HGBL, NOS.63,64 Additionally, the Nordic Lymphoma Group NLG-LBC-06 trial used DA-EPOCH-R therapy for aggressive B-cell lymphomas with MYC-R, TP53 deletion or p53 overexpression, DEL, or DHL status.65 However, considering the lack of control groups in these trials, lack of prognostic significance of MYC-R in DLBCL, and inconsistent results from retrospective series of HGBL/BCLU, the significance of a single-hit MYC-R for treatment selection in HGBL, NOS, remains uncertain.59,66 Correlative studies are needed to delineate the prognostic and predictive value of molecular markers, including MYC translocations, amplifications, and hotspot mutations, TP53 or CDKN2A/B alterations, complex karyotype, and GEP signatures. The morphologic and molecular heterogeneities of HGBL, NOS, are likely also reflected in its responsiveness to therapy.
Clinical trial outcomes of adults with HGBL, NOS
| Study | Treatment | Study population* | HGBL, NOS, n | PFS, % (y)† | OS, % (y)† |
|---|---|---|---|---|---|
| Cook et al, 61 | R-CHOP ± ASCT | DLBCL or HGBL | 31 | 69 (2) | 77 (2) |
| Persky et al, 71 | R-CHOP ± IFRT/IbT | Localized DLBL or HGBL | 22 | 87 (5)‡ | 89 (5)‡ |
| Mead et al, 76 | CODOX-M/IVAC | BL or DLBCL with Ki-67 >95% | NR (DLBCL, 57) | 55 (2) | 59 (2) |
| Corazzelli et al, 77 | R-CODOX-M/IVAC | BL or HGBL | 15 | 65 (4) | NR |
| McMillan et al, 82 | R-CODOX-M/IVAC | DLBCL with IPI 3-5 | NR | 68 (2) | 76 (2) |
| Dunleavy et al, 63 | DA-EPOCH-R | DLBCL with MYC-R | 10 | 71 (4) | 77 (4) |
| Hoelzer et al, 85 | GMALL-B-ALL/NHL2002 | BL, HGBL, Burkitt leukemia | NR | 75 (5)‡ | 80 (5)‡ |
| Thomas et al, 91 | R-hyperCVAD/MA | BL, HGBL, B-ALL | 16 | 80 (3)‡ | 89 (3)‡ |
| Rizzieri et al, 90 | CALGB 10002 | BL or HGBL | 25 | 64 (2) | 64 (2) |
| Study | Treatment | Study population* | HGBL, NOS, n | PFS, % (y)† | OS, % (y)† |
|---|---|---|---|---|---|
| Cook et al, 61 | R-CHOP ± ASCT | DLBCL or HGBL | 31 | 69 (2) | 77 (2) |
| Persky et al, 71 | R-CHOP ± IFRT/IbT | Localized DLBL or HGBL | 22 | 87 (5)‡ | 89 (5)‡ |
| Mead et al, 76 | CODOX-M/IVAC | BL or DLBCL with Ki-67 >95% | NR (DLBCL, 57) | 55 (2) | 59 (2) |
| Corazzelli et al, 77 | R-CODOX-M/IVAC | BL or HGBL | 15 | 65 (4) | NR |
| McMillan et al, 82 | R-CODOX-M/IVAC | DLBCL with IPI 3-5 | NR | 68 (2) | 76 (2) |
| Dunleavy et al, 63 | DA-EPOCH-R | DLBCL with MYC-R | 10 | 71 (4) | 77 (4) |
| Hoelzer et al, 85 | GMALL-B-ALL/NHL2002 | BL, HGBL, Burkitt leukemia | NR | 75 (5)‡ | 80 (5)‡ |
| Thomas et al, 91 | R-hyperCVAD/MA | BL, HGBL, B-ALL | 16 | 80 (3)‡ | 89 (3)‡ |
| Rizzieri et al, 90 | CALGB 10002 | BL or HGBL | 25 | 64 (2) | 64 (2) |
ASCT, autologous stem cell transplantation; B-ALL, B-cell acute lymphoblastic leukemia; CALGB, Cancer and Leukemia Group B; GMALL, German Multicenter Study Group for Adult Acute Lymphoblastic Leukemia; IFRT/IbT, involved-field radiation therapy followed by ibritumomab tiuxetan consolidation; NR, not reported; R-hyperCVAD/MA, rituximab plus fractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone alternating with high-dose methotrexate and cytarabine.
In this table, the term HGBL encompasses Burkitt-like lymphoma, BCLU, DHL, and HGBL, NOS.
Outcomes of the HGBL subset, unless stated otherwise.
Outcomes in the entire study population (results reportedly did not significantly differ for HGBL).
Is R-CHOP adequate for HGBL, NOS?
The main dilemma in HGBL, NOS, is whether DLBCL-like therapy is sufficient, or whether intensified regimens used for BL/DHL are necessary. This area lacks prospective, histology-specific data. Outcomes of HGBCL, NOS, are nearly impossible to disentangle from historical series that variably combined BCLU, DHL, DEL, and DLBCL with MYC-R or even MYC amplification. In the prerituximab era, outcomes of Burkitt-like lymphoma or BCLU in trials seemed similar to those of DLBCL, but MYC-R was associated with poor survival.61,67,68 Retrospective studies do not paint a consistent picture and are subject to bias in patient and treatment selection. One series of 33 pediatric and adult patients with BL or Burkitt-like lymphomas noted that adults without MYC-R who received low-intensity regimens had significantly worse survival compared with those treated with BL protocols because of MYC-R. This difference was absent among children uniformly treated with intensive chemotherapy.66 In contrast, in a series of 52 patients with BCLU treated with R-CHOP or R-hyperCVAD/MA, prognosis was significantly worse for patients with MYC-rearranged tumors who received R-CHOP, but for those without MYC-R, there was no difference in prognosis between the regimens.59 Perry et al58 reported 39 BCLU cases (68% of which would be now classified as HGBL, NOS, and 32% as DHL by FISH) treated mostly with R-CHOP. Only 43% of patients attained a CR, and in most, the disease relapsed soon after completion of chemotherapy (including 35% in the CNS). Median OS was 9 months, with only 30% OS at 5 years and no difference by DHL or MYC-R status. In the aforementioned Chinese case series of HGBL, NOS, survival was significantly worse after R-CHOP compared with more intensive regimens.29 These collective data suggest that, as in the case of DHL, R-CHOP is likely suboptimal for many patients with advanced-stage HGBL, NOS, and we favor intensified regimens, although we acknowledge the uncertainty (Figure 4).
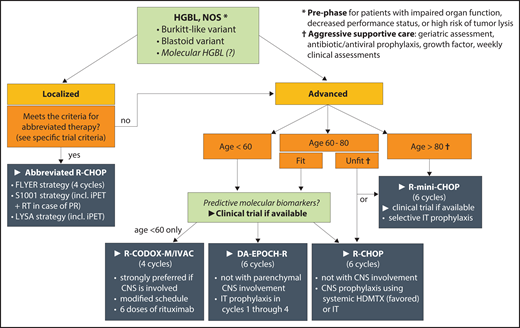
First-line therapy for HGBL, NOS. Recommendations are based on available experience in aggressive lymphomas and the authors’ preferences, because histology-specific data are sparse; areas of priority research are italicized. HDMTX, high-dose methotrexate; iPET, interim positron emission tomography; IT, intrathecal; PR, partial response; RT, radiation therapy.
First-line therapy for HGBL, NOS. Recommendations are based on available experience in aggressive lymphomas and the authors’ preferences, because histology-specific data are sparse; areas of priority research are italicized. HDMTX, high-dose methotrexate; iPET, interim positron emission tomography; IT, intrathecal; PR, partial response; RT, radiation therapy.
In contrast, for the infrequent cases of early-stage HGBL, NOS, outcomes with R-CHOP are similar to those in DLBCL. One retrospective series of early-stage MYC-rearranged DLBCL/HGBL (50% DHL) found no difference in survival between R-CHOP and intensified regimens.69 Seventeen HGBL, NOS, or Burkitt-like tumors were included in the FLYER trial, which enrolled patients with aggressive B-cell lymphomas with no IPI risk factors and tumors <7.5 cm.70 PFS at 3 years was 96% with 4 cycles of R-CHOP plus 2 doses of rituximab, and no patient with HGBL relapsed. In the S1001 trial, 17% of 132 enrolled patients had HGBL, NOS.71 In this phase 2 trial, patients underwent positron emission tomography after 3 cycles of R-CHOP, and those with a CR (89%) received 1 additional R-CHOP cycle, whereas others received consolidative radiation therapy and radioimmunotherapy. Aggregate 5-year PFS was 87%. Recently, the phase 3 LYSA LNH 09-1B trial reported noninferiority of 4 vs 6 R-CHOP cycles in early-stage aggressive B-cell lymphoma with age-adjusted IPI of 0, also guided by interim positron emission tomography response, and including 16 HGBL cases.72 On the basis of these data, the FLYER, S1001, and LYSA strategies can be considered for early-stage HGBL, NOS, without additional risk factors.
For older or unfit patients with HGBL, NOS, we favor dose-attenuated R-mini-CHOP (rituximab plus reduced-dose CHOP) as a standard approach, guided by geriatric assessment, but recommend clinical trials investigating incorporation of novel targeted approaches whenever available (eg, R-mini-CHOP with azacitidine; registered at www.clinicaltrials.gov as #NCT04799275).73,74
Intensified therapy for HGBL, NOS
R-CODOX-M/IVAC
The dose-intense R-CODOX-M/IVAC regimen was pioneered by Magrath et al75 for younger patients with BL or lymphoblastic lymphoma.31,76 R-CODOX-M/IVAC has been applied in BCLU,77,78 HIV-associated BL,79-81 and high-risk DLBCL.82 In the LY-10 trial, 57 patients with aggressive DLBCL/HGBL achieved 55% PFS and 59% OS at 2 years after CODOX-M/IVAC without rituximab.76 In a recent phase 2 trial using modified CODOX-M/IVAC plus 8 doses of rituximab for DLBCL/HGBL, 2-year PFS was 68%, including patients with baseline CNS involvement (Table 1).82 Toxicities were significant, with grade 4 neutropenia in 88%, thrombocytopenia in 61%, grade ≥3 infections in 71%, and treatment-related mortality of 4% (concentrated among patients who were age >50 years and had poor performance status). Real-world data in BL indicate treatment-related mortality of 5%, mainly resulting from sepsis.31 R-CODOX-M/IVAC has been further modified to ameliorate its toxicity, and we recommend the adjusted schedule, with 6 doses of rituximab.78,82,83 Absent randomized data, R-CODOX-M/IVAC may be preferable for patients age <50 to 60 years and with CNS involvement.
DA-EPOCH-R
DA-EPOCH-R is an intermediate-intensity regimen for high-grade lymphomas including BL and DHL.84 In standard DLBCL, it did not demonstrate an advantage over R-CHOP, while significantly increasing rates of febrile neutropenia (35%) and grade ≥3 peripheral neuropathy (19%).13 Nevertheless, in a phase 2 trial focused on lymphomas with MYC-R, 4-year event-free survival after DA-EPOCH-R in HGBL (HGBL, NOS, n = 10; DHL, n = 24) was 71% (Table 2).63 The study enrolled patients up to age 80 years, indicating that DA-EPOCH-R is an important option for older patients with HGBL, NOS, because administration of other intensified regimens at age >60 years is unsafe. However, even this regimen may be too aggressive for patients age >80 years (with or without comorbidities), because in this group the risk/benefit ratio of full-dose chemotherapy becomes uncertain.
Prephase
The use of prephase therapy to improve a patient’s performance status before the start of definitive chemotherapy has not been specifically studied in HGBL. However, prephase therapy with corticosteroid with or without vincristine and/or fractionated cyclophosphamide is commonly used in BL.81,85,86 In DLBCL, the use of corticosteroid prephase at least 1 week before the start of chemotherapy improved performance status and reduced the number of treatment-associated deaths.87
CNS prophylaxis
Unlike in BL, which shows predominantly leptomeningeal CNS involvement, parenchymal CNS relapses are more common in HGBL and DLBCL.57,60,88 Systemic prophylaxis using high-dose methotrexate is included in R-CODOX-M/IVAC and can be incorporated into R-CHOP. For HGBL, we favor early intercalated administration when feasible.89 In the phase 2 DA-EPOCH-R trial, CNS prophylaxis comprised 8 administrations of intrathecal methotrexate on days 1 and 5 during cycles 3 through 6.63 Data from BL indicate that this schedule may not be rigorously followed, even in academic centers, thus compromising CNS-related outcomes.88 The phase 3 trial included 4 intrathecal injections delivered during cycles 3 to 6 of DA-EPOCH-R.13 Although the efficacy of intrathecal prophylaxis against parenchymal CNS recurrence is uncertain, in HGBL we favor earlier administration (whenever possible, during a staging lumbar puncture, and regardless of clinical risk factors), with additional doses delivered during cycles 2 through 4. Any benefit of high-dose methotrexate consolidation after DA-EPOCH-R is uncertain, so when a compelling indication for systemic CNS prophylaxis exists, we favor R-CODOX-M/IVAC, which assures early delivery.
Other dose-intense regimens have been studied in BL and HGBL/BCLU with comparable outcomes, but they do not offer conceptual advantages over R-CODOX-M/IVAC.85,90,91 The R-hyperCVAD/MA regimen is included in the National Comprehensive Cancer Network guidelines, but it requires prolonged therapy (8 cycles) and may be associated with a higher treatment-related mortality.31
Autologous transplantation for consolidation
Consolidation using high-dose chemotherapy and ASCT in first remission is a tempting approach in high-grade lymphomas, which have poor prognosis after chemotherapy alone. However, despite 4 randomized trials, it has not resulted in improved OS, even though some trials showed a PFS advantage in DLBCL with intermediate or high IPI.14-17 These trials did not focus on HGBL, diluting any potential benefit. A large retrospective analysis of 159 DHLs did not show a benefit of consolidative ASCT in first CR.92 Acknowledging the uncertainty related to data extrapolation, at present there is no evidence that ASCT improves outcomes for patients with HGBL, NOS, who attain CR with chemotherapy.
Relapsed/refractory HGBL
Salvage chemotherapy and transplantation
Data on treatment or outcomes of recurrent HGBL, NOS, are scant. Traditional approaches, modeled after DLBCL, attempt to reinduce remission and consolidate with ASCT. In the randomized CORAL trial, MYC-R was noted in a minority of patients and was notably associated with 4-year PFS of only 18%; outcomes were even worse (2-year OS, 0% to 10%) in observational studies.93,94 Among 267 patients with aggressive B-cell lymphomas (44% HGBL/BCLU) relapsing after intensive immunochemotherapy, 43% responded to salvage chemotherapy, 23% had a CR, and 15% proceeded to ASCT, achieving 3-year PFS of 42%; PFS for all patients was 14%.95 In a retrospective analysis of relapsed DHL, survival seemed better with salvage allogeneic stem cell transplantation (4-year PFS, 40%; OS, 50%).96 With regard to recently approved options for relapsed/refractory DLBCL, no confirmed cases of HGBL were reported in the registration trial of polatuzumab vedotin97; 1 patient with DHL responded to selinexor98; 4 of 7 patients with MYC-rearranged lymphomas (including 2 DHLs) responded to tafasitamab plus lenalidomide,99 and 22% of those with DHL responded to loncastuximab tesirine.100
T cell–directed immunotherapy
A handful of patients with HGBL, NOS, have participated in trials of CD19-directed chimeric antigen receptor T-cell therapy, with promising results. Nineteen patients with confirmed DHL enrolled in the JULIET trial (tisagenlecleucel) and showed overall response rate of 50%, similar to DLBCL (52%).101 In ZUMA-1 (axicabtagene ciloleucel), 7 patients had HGBL, including 2 with HGBL, NOS; 1 achieved a CR, and 1 a partial response.102 The TRANSCEND-NHL-001 trial of lisocabtagene maraleucel included 36 patients with DHL whose response rate was similar to those with DLBCL (76% and 68%, respectively).103 In 1 study describing real-world outcomes of axicabtagene ciloleucel, 12-month PFS was 39% in HGBL, and OS was 69%, neither significantly different from those in DLBCL.104 A second real-world study identified 17 patients with HGBL, NOS, of whom 15 (88%) responded to chimeric antigen receptor T cells.105
These responses to T cell–directed immunotherapy are encouraging in HGBL, which is characterized by downregulation of MHC genes and immune response pathways, low expression of PD-L1 on malignant B cells, and low macrophage content in the microenvironment.43,44,106-108 Efficacy of other forms of immunotherapy in HGBL remains to be determined. Anecdotal responses to blinatumomab (n = 1),109 CD20/CD3-bispecific antibody mosunetuzumab (n = 2),110 and anti-CD47 antibody Hu5F9-G4 (n = 1) have been reported.111
Future directions
HGBL, NOS, as outlined in the 2016 WHO classification, is a poorly defined and highly heterogeneous entity. Delineation of aggressive high-grade lymphomas that require treatment other than R-CHOP remains a critical priority for research. This goal will likely require replacing the cytomorphologic definition of HGBL, NOS, with more objective GEP or genomic criteria that can account for the biologic similarity between DHL (as identified by FISH) and molecular HGBL, which encompasses >50% of HGBLs, NOS, and ∼15% of DLBCLs, NOS. Successful translation of this knowledge depends on deployment of rapid, readily available, and affordable molecular diagnostics in clinical practice. Examples of studies that can pave the way for future treatment of HGBL include the Alliance A051701 phase 2/3 trial of chemotherapy with or without venetoclax in DEL/DHL (registered at www.clinicaltrials.gov as #NCT03984448) and the ZUMA-12 trial of axicabtagene ciloleucel for patients with DHL or DLBCL who do not achieve early metabolic CR with chemotherapy (registered at www.clinicaltrials.gov as #NCT03761056). Genomic analyses will be important to delineate benefits of these treatments for HGBL. Apart from immunotherapeutic approaches, other potential strategies include epigenetic modifiers, phosphoinositide 3-kinase ihibitors,42,112 bromodomain inhibitors,113 novel immunomodulatory agents, and antibody-drug conjugates.100 Application of these therapies will need to be refined in the context of tumor biology, considering the highly proliferative nature of HGBL and the molecular mechanisms that drive it. After treating unclassifiable tumors as oddities, with diagnoses of exclusion, the prospect of improving outcomes via refined molecular diagnostics and targeted therapeutic approaches for these aggressive B-cell lymphomas is potentially achievable.
Authorship
Contribution: A.J.O., H.K., and A.M.E. wrote the manuscript and approved the final version of the manuscript.
Conflict-of-interest disclosure: A.J.O. reports research funding from Genentech, Adaptive Biotechnologies, Acrotech Biopharma, Celldex Therapeutics, TG Therapeutics, and Precision BioSciences. A.M.E. reports research funding from ORIEN, Leukemia & Lymphoma Society, National Institutes of Health, and National Cancer Institute; has served on advisory boards (research related) for Seattle Genetics, MorphoSys, Karyopharm, Novartis, Pharmayclics, AbbVie, and Epizyme; and has served as consultant (educational related) for Research to Practice, Curio, Cota, Patient Power, and OncLive. H.K. reports no competing financial interests.
Correspondence: Andrew M. Evens, Rutgers Cancer Institute of New Jersey, 195 Little Albany St, New Brunswick, NJ 08901; e-mail: andrew.evens@rutgers.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal