

Key Points
CD19.22.BBζ CAR T cells were well tolerated and effective in pediatric B-ALL, but persistence and CD22 targeting were limited.
A novel bicistronic CD19.28ζ/CD22.BBζ CAR T-cell enhanced dual-targeting efficacy and cytokine production in preclinical models.

Abstract
Remission durability following single-antigen targeted chimeric antigen receptor (CAR) T-cells is limited by antigen modulation, which may be overcome with combinatorial targeting. Building upon our experiences targeting CD19 and CD22 in B-cell acute lymphoblastic leukemia (B-ALL), we report on our phase 1 dose-escalation study of a novel murine stem cell virus (MSCV)-CD19/CD22-4-1BB bivalent CAR T-cell (CD19.22.BBζ) for children and young adults (CAYA) with B-cell malignancies. Primary objectives included toxicity and dose finding. Secondary objectives included response rates and relapse-free survival (RFS). Biologic correlatives included laboratory investigations, CAR T-cell expansion and cytokine profiling. Twenty patients, ages 5.4 to 34.6 years, with B-ALL received CD19.22.BBζ. The complete response (CR) rate was 60% (12 of 20) in the full cohort and 71.4% (10 of 14) in CAR-naïve patients. Ten (50%) developed cytokine release syndrome (CRS), with 3 (15%) having ≥ grade 3 CRS and only 1 experiencing neurotoxicity (grade 3). The 6- and 12-month RFS in those achieving CR was 80.8% (95% confidence interval [CI]: 42.4%-94.9%) and 57.7% (95% CI: 22.1%-81.9%), respectively. Limited CAR T-cell expansion and persistence of MSCV-CD19.22.BBζ compared with EF1α-CD22.BBζ prompted laboratory investigations comparing EF1α vs MSCV promoters, which did not reveal major differences. Limited CD22 targeting with CD19.22.BBζ, as evaluated by ex vivo cytokine secretion and leukemia eradication in humanized mice, led to development of a novel bicistronic CD19.28ζ/CD22.BBζ construct with enhanced cytokine production against CD22. With demonstrated safety and efficacy of CD19.22.BBζ in a heavily pretreated CAYA B-ALL cohort, further optimization of combinatorial antigen targeting serves to overcome identified limitations (www.clinicaltrials.gov #NCT03448393).
Introduction
Despite tremendous efficacy of single-antigen targeted chimeric antigen receptor (CAR) T cells for B-cell acute lymphoblastic leukemia (B-ALL),1-8 leukemic antigen modulation remains a major mechanism of resistance and treatment failure.9 Simultaneous multi-antigen CAR T-cell targeting may reduce antigen-negative relapse and extend remission durability. Building upon our institutional experience with single-antigen CD194,5 and CD22,10,11 CAR T-cell trials, we developed a bivalent CD19/CD22 CAR T-cell construct (CD19.22.BBζ) incorporating both single chain variable fragments (scFvs) into a tandem construct with a loop configuration using a 4-1BB co-stimulatory domain.12 The potential advantage of bivalent CARs includes the relative ease and cost of manufacturing a single CAR T-cell product using 1 vector that uniformly expresses both scFvs.13-16 Alternative dual-antigen models include co-infusion, which is limited by logistical and practical considerations of manufacturing 2 separate products,17,18 or co-transduction strategies, where skewed transduction and/or expansion of 1 CAR T-cell product may preclude optimal bispecific targeting.12,19,20
We tested this novel construct in a phase 1 dose-escalation study in children and young adults (CAYA) with relapsed/refractory (r/r) B-ALL and demonstrate manufacturing feasibility, a well-tolerated toxicity profile, and clinical efficacy. Unexpectedly, despite incorporation of a 4-1BB co-stimulatory domain, which has generally been associated with persistence of CD19 CAR and CD22 CAR T cells,1,21 CAR T-cell expansion, persistence, and CD22 targeting were limited in comparison with our single-antigen targeted CD22.BBζ CAR T-cell, prompting further investigation of critical properties of this CD19.22.BBζ construct. Development of a novel bicistronic CD19.28ζ/CD22.BBζ CAR T-cell construct to improve on limitations of our current construct was subsequently pursued.
Methods
Participants and study design
This single-center, phase 1 dose-escalation study testing CD19.22.BBζ in CAYA with CD19+/CD22+ B-cell malignancies in the Pediatric Oncology Branch, National Cancer Institute (NCI) included patients aged 3 to 35 years with r/r disease, adequate organ function, and performance status ≥ 50%. Inclusion required CD19+ expression on >90% of malignant cells by flow cytometry (FC; or >15% by immunohistochemistry for lymphomatous disease) and any CD22 positivity. Patients receiving prior CAR T-cell therapy (CAR-pretreated) were eligible if they were ≥30 days from last infusion and circulating levels of genetically modified T cells were <5% by FC. Importantly, patients who were CAR-pretreated receiving interim allogeneic hematopoietic stem cell transplant (HSCT) were categorized as CAR-naïve. Central nervous system 3 (CNS3) disease was exclusionary. The protocol was approved by the NCI Institutional Review Board and registered at clinicaltrials.gov (#NCT03448393). This report incorporates data from all patients treated on-study from May 2018 through August 2021, with a data cutoff of September 30, 2021. The primary objective was to evaluate safety and toxicity. Secondary objectives included manufacturing feasibility and clinical response. Additional details are in the supplemental Appendix available on the Blood Web site.
Three dose levels (DLs), measured by transduced CAR T-cells/kg, included: DL1 (3 × 105), DL2 (1 × 106), and DL3 (3 × 106). All received standard lymphodepletion (LD) with fludarabine 25 mg/m2 days −4 to −2 and cyclophosphamide 900 mg/m2 on day −2 (n = 18) or intensified LD after a protocol amendment for patients who were CAR-pretreated (fludarabine, 30 mg/m2, days −5 to −2, and cyclophosphamide, 600 mg/m2 on days −3 and −2; n = 2). For patients who were HSCT-naïve and achieved remission following CAR T-cell infusion, consolidative HSCT was recommended, but not required as per standard-of-care in high-risk ALL.22
Manufacturing of CAR T cells
CAR T-cells were manufactured (typically within 7 days) on the CliniMACS Prodigy closed system device using a CD19.22.BBζ bivalent CAR vector, comprised of FMC63 murine scFv (anti-CD19) and m971 human scFv (anti-CD22) under control of the murine stem cell virus (MSCV) promoter and with interleukin 2 (IL-2) support. Following successful manufacture of the first product (albeit with limited in vitro expansion), the second product failed manufacturing. A minor modification (washout of activation beads and lentiviral vector at day 3 instead of day 5) led to successful manufacture using the same apheresis material. The modified protocol was used for all subsequent products.23
Assessment of response
Disease assessments were performed pre-LD (baseline) and at day 28 ± 4 days. Patients with FC detectable CNS1 status were classified as CNS1 FCpositive. Clinically indicated fluorodeoxyglucose-positron emission tomography scan was performed in patients with non-Hodgkin lymphoma (NHL) and for assessment of known or suspected extramedullary disease (EMD).24
Overall best response incorporated bone marrow (BM), cerebrospinal fluid, and/or fluorodeoxyglucose-positron emission tomography scan assessments at day 28. Complete response (CR) was defined by the absence of disease in any compartment. Discrepant disease responses (eg, BM CR with residual EMD) were categorized by the overall worst response.
Toxicity assessments
Adverse events (AEs) were graded using Common Terminology Criteria for Adverse Events through day 28 after infusion or resolution to baseline. Cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) were graded using the American Society for Transplantation and Cellular Therapy consensus criteria.25 Cytopenias, infection, and B-cell aplasia were additionally captured. An NCI-developed caregiver-based neuro-symptom checklist (NSC)26,27 was administered at protocol-specified time points.
Clinical correlative studies
Neurocognitive testing
Psychologists administered a brief neurocognitive battery (including the Cogstate computerized test), assessing attention, processing speed, working and visual learning memory, executive function, and verbal fluency pre- and post-CD19.22.BBζ CAR T cells (once between days 21 and 28).
Biologic correlative studies
CD19.22.BBζ CAR T-cell expansion and persistence by FC was assessed weekly for the first month, at 2 months, and then every 3 to 6 months as feasible. Cytokines, absolute lymphocyte counts, and human–anti-mouse antibodies (HAMAs) were evaluated serially during the first month.
CD19, CD22, and CD19/22 comparison studies
Comparison studies between patients receiving CD19.28ζ, CD22.BBζ, and CD19.22.BBζ constructs included evaluation of CAR T-cell expansion and persistence, serum cytokines, C-reactive protein (CRP), and ferritin. Manufacturing methods and results from our CD19.28ζ and CD22.BBζ trials were previously reported.5,10,11,28
Reverse translational investigations
Comparison of CAR efficacy as a function of the promoter
To interrogate the role of the promoter, we compared our MSCV-CD19.22.BBζ construct to an EF1α-CD19.22.BBζ CAR, using identical manufacturing methodologies for vector and CAR T-cell production.12
For evaluation of CAR function, transduced T cells were cocultured ex vivo with human B-ALL NALM6 cell lines (CD19+CD22+), as well as with engineered CD19+CD22neg, CD19negCD22+, and CD19negCD22neg NALM6. For ex vivo cytokine analyses, cells were cocultured at a 1:1 E/T ratio on a TECAN robotic platform programmed to perform automatic collection and replenishment of supernatants at frequent time points.29 Collected supernatants were evaluated by cytokine bead array (BD Biosciences, San Diego, CA) and run on a Fortessa FACS cytometer (BD Biosciences) to measure serial cytokine concentrations.
The ability of MSCV-CD19.22.BBζ and EF1α-CD19.22.BBζ CAR to inhibit ALL proliferation in vivo was assessed in NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ; Jackson Laboratories) mice using bioluminescent NALM6 cell lines.12 Animal experiments were carried out under protocols approved by the NCI Bethesda Animal Care and Use Committee. The indicated NALM6 cell lines (1e6) were injected 3 days before adoptive transfer of transduced T-cells (3e6). For evaluation of leukemia growth, mice were injected intraperitoneally with d-luciferin (3 mg, Caliper Life Sciences) and imaged with an exposure time of 1 minute. Living Image Version 4.1 software (Caliper Life Sciences) was used to analyze bioluminescent signal flux for each mouse.
Generation of bicistronic CD19/CD22 CAR constructs
Novel bicistronic EF1α constructs harboring CD22 and CD19 CARs containing both CD28 and 41BB costimulatory domains were generated, with a 2A peptide from porcine teschovirus-1 and an upstream furin recognition incorporated to allow self-cleavage and removal of 2A residues from the upstream gene, respectively.30 In constructs harboring 4-1BB endodomains, the hinge and transmembrane domain were derived from CD8, whereas constructs harboring CD28 costimulatory domains contained CD28 hinge and transmembrane domains.
Statistical analysis
Fisher’s exact test was used to compare binary outcomes between 2 groups. A Mann-Whitney test was used to compare continuous variables between 2 groups. Kaplan-Meier survival curves were used to estimate overall survival (OS) and relapse-free survival (RFS), the latter restricted to those who achieved a CR. OS was calculated from the date of CAR infusion until the date of death or last follow-up with a cutoff of 30 September 2021. RFS was calculated from CAR infusion until the date of relapse or last follow-up without censoring for HSCT. Statistical analyses were performed using GraphPad Prism version 9.0 or SAS version 9.4. See supplemental Appendix for statistical analyses of neurocognitive testing and laboratory investigations.
Results
Patient demographics
Twenty-one of 22 enrolled patients received CD19.22.BBζ CAR T cells (Table 1). The 20 patients with B-ALL comprise the primary analysis cohort. One patient was not infused because of progressive leukemia/concurrent infection. One patient with NHL is described in the supplemental Appendix. All had a product successfully manufactured (supplemental Table 1; supplemental Figure 1A). The median age was 20.5 years (range: 5.4-34.6 years). Twelve (60%) had relapsed after at least 1 prior allogeneic HSCT, and 15 (75%) had received either CD19- or CD22-directed immunotherapy. Nine (45%) had high-disease burden (≥5% blasts), and all were CNS1, 3 with CNS1 FCpositive. Eight (40%) had non-CNS extramedullary disease.
Demographics of patients with B-ALL
| Characteristics | Subjects treated (n = 20) | CAR naïve (n = 14) | CAR pretreated (n = 6) | |
|---|---|---|---|---|
| Age, median (y), range | 20.5 (5.4-34.6) | 25.2 (5.4-34.6) | 19.1 (8.3-23.8) | |
| Sex | ||||
| Male | 12 (60%) | 9 (64.3%) | 3 (50%) | |
| Female | 8 (40%) | 5 (35.7%) | 3 (50%) | |
| Disease status and prior therapy | ||||
| 2-3 previous lines of therapy | 8 (40%) | 8 (57.1%) | 0 | |
| >4 previous lines of therapy | 12 (60%) | 6 (42.9%) | 6 (100%) | |
| No. of prior allogeneic transplants | ||||
| 0 (HSCT naïve) | 8 (40%) | 7 (50%) | 1 (16.7%) | |
| ≥1 | 12 (60%) | 7 (50%) | 5 (83.3%) | |
| Prior immunotherapy | ||||
| Prior CD19 targeted therapy* | 15 (75%) | 9 (64.3%) | 6 (100%) | |
| Type of CD19 targeted therapy | ||||
| Prior blinatumomab | 14 (70%) | 8 (57.1%) | 6 (100%) | |
| Prior CD19 CAR T cells | 7 (35%) | 2 (14.3%)† | 5 (83.3%)‡ | |
| Prior CD22 targeted therapy§ | 6 (30%) | 1 (7.1%) | 5 (83.3%) | |
| Type of CD22 targeted therapy | ||||
| Prior inotuzumab | 5 (25%) | 1 (7.1%) | 4 (66.7%) | |
| Prior CD22 CAR T cells | 2 (10%) | 0 | 2 (33.3%)‡ | |
| Marrow disease burden at initiation of LD | ||||
| MRD negativeǁ | 1 (5%) | 1 (7.1%) | 0 | |
| M1 | 10 (50%) | 7 (50%) | 3 (50%) | |
| ≥M2 | 9 (45%) | 6 (42.9%) | 3 (50%) | |
| CNS disease status at initiation of LD | ||||
| CNS1 | 17 (85%) | 12 (85,7%) | 5 (83.3%) | |
| CNS1 with flow + disease | 3 (15%) | 2 (14.3%) | 1 (16.7%) | |
| Extramedullary disease status at initiation of LD | ||||
| Extramedullary disease (Non-CNS) at treatment | 8 (40%) | 6 (42.8%) | 2 (33.3%) | |
| Characteristics | Subjects treated (n = 20) | CAR naïve (n = 14) | CAR pretreated (n = 6) | |
|---|---|---|---|---|
| Age, median (y), range | 20.5 (5.4-34.6) | 25.2 (5.4-34.6) | 19.1 (8.3-23.8) | |
| Sex | ||||
| Male | 12 (60%) | 9 (64.3%) | 3 (50%) | |
| Female | 8 (40%) | 5 (35.7%) | 3 (50%) | |
| Disease status and prior therapy | ||||
| 2-3 previous lines of therapy | 8 (40%) | 8 (57.1%) | 0 | |
| >4 previous lines of therapy | 12 (60%) | 6 (42.9%) | 6 (100%) | |
| No. of prior allogeneic transplants | ||||
| 0 (HSCT naïve) | 8 (40%) | 7 (50%) | 1 (16.7%) | |
| ≥1 | 12 (60%) | 7 (50%) | 5 (83.3%) | |
| Prior immunotherapy | ||||
| Prior CD19 targeted therapy* | 15 (75%) | 9 (64.3%) | 6 (100%) | |
| Type of CD19 targeted therapy | ||||
| Prior blinatumomab | 14 (70%) | 8 (57.1%) | 6 (100%) | |
| Prior CD19 CAR T cells | 7 (35%) | 2 (14.3%)† | 5 (83.3%)‡ | |
| Prior CD22 targeted therapy§ | 6 (30%) | 1 (7.1%) | 5 (83.3%) | |
| Type of CD22 targeted therapy | ||||
| Prior inotuzumab | 5 (25%) | 1 (7.1%) | 4 (66.7%) | |
| Prior CD22 CAR T cells | 2 (10%) | 0 | 2 (33.3%)‡ | |
| Marrow disease burden at initiation of LD | ||||
| MRD negativeǁ | 1 (5%) | 1 (7.1%) | 0 | |
| M1 | 10 (50%) | 7 (50%) | 3 (50%) | |
| ≥M2 | 9 (45%) | 6 (42.9%) | 3 (50%) | |
| CNS disease status at initiation of LD | ||||
| CNS1 | 17 (85%) | 12 (85,7%) | 5 (83.3%) | |
| CNS1 with flow + disease | 3 (15%) | 2 (14.3%) | 1 (16.7%) | |
| Extramedullary disease status at initiation of LD | ||||
| Extramedullary disease (Non-CNS) at treatment | 8 (40%) | 6 (42.8%) | 2 (33.3%) | |
Data presented as n (%) unless otherwise indicated. Definitions: M1: <5% blasts in the bone marrow; M2: 5%-25% blasts in bone marrow; M3: >25% blasts in bone marrow. CNS1: No blasts on cytospin; CNS1 + flow cytometry positive: No blasts on cytospin however blasts detected by multiparametric flow cytometry.
Five subjects received both CD19 CAR and blinatumomab.
Two subjects received prior CD19 CAR and had an interval HSCT thus are considered CAR naïve.
One patient received prior CAR treatment with both a CD19 and a CD19/CD22 CAR T-cell infusion before receipt of this CAR construct.
One subject received prior CD22 CAR and inotuzumab.
One subject with ALL did not have bone marrow involvement, however, had extramedullary disease.
Six patients (30%) were CAR pretreated (prior CD19 CAR [n = 5], prior CD22 CAR [n = 1], 1 patient received both CD19-targeted and an alternate CD19/22 CAR T-cell). Prior CAR T-cell exposure occurred at a median of 351 days (range: 82-767 days) before CD19.22.BBζ CAR T-cell infusion.
Toxicity
CRS and ICANS
Ten patients (50%) developed CRS (7 with grade 1-2; 3 with grade 3; Table 2) at a median of 4.5 days after infusion (range: 2-9 days). Those with grade 3 CRS had high-disease burden and required transient intensive care support for hypotension. Six received tocilizumab, 3 received concomitant corticosteroids (for persistent fevers [n = 2] and disseminated intravascular coagulation [n = 1]). One patient developed ICANS on day +7 (grade 3 with acute-onset aphasia, right-sided weakness, agitation, and confusion) and constituted a dose-limiting toxicity. IV dexamethasone, followed by a single dose of intrathecal (IT) hydrocortisone (15 mg), was used for further management. Within hours of receiving IT hydrocortisone, ICANS improved to grade 1 and fully resolved within 48 hours. Hemophagocytic lymphohistiocytosis-like toxicities, as seen with CD22 CAR T cells,31 were not experienced (supplemental Table 2 for research related ≥ grade 3 adverse effects).
Toxicity, CRS management, and response profile in patients with B-ALL
| Category | Sub-category | Total subjects treated (n = 20) | CAR naïve (n = 14) | CAR pretreated (n = 6) |
|---|---|---|---|---|
| Dose level | Dose level 1 (3 × 105 CAR T cells/kg) | 4 (20%) | 1 (7.1%) | 3 (50%) |
| Dose level 2 (1 × 106 CAR T cells/kg) | 4 (20%) | 3 (21.4%) | 1 (16.7%) | |
| Dose level 3 (3 × 106 CAR T cells/kg) | 12 (60%) | 10 (71.4%) | 2 (33.3%) | |
| LD | Standard LD | 18 (90%) | 14 (100%) | 4 (66.7%) |
| Intensified LD | 2 (10%) | 0 | 2 (33.3%) | |
| CRS and ICANS* | No CRS | 10 (50%) | 5 (35.7%) | 5 (83.3%) |
| CRS grade 1-2 | 7 (35%) | 6 (42.9%) | 1 (16.7%) | |
| CRS grade 3-4 | 3 (15%) | 3 (21.4%) | 0 | |
| ICANS | 1 (5%) | 0 | 1 (16.7%) | |
| Treatment | Tocilizumab treatment | 6 (30%) | 5 (35.7%) | 1 (16.7%) |
| Corticosteroids treatment† | 4 (20%) | 3 (21.4%) | 1 (16.7%) | |
| Bone marrow response‡ | MRD negative CR | 16 (80%) | 12 (85.7%) | 4 (66.7%) |
| Partial response | 1 (5%) | 1 (7.1%) | 0 | |
| Stable/progressive disease | 3 (15%) | 1 (7.1%) | 2 (33.3%) | |
| Extramedullary response (non-CNS) (n = 8)§ | Complete response | 3 (37.5%) | 3 (50%) | 0 |
| Partial response | 3 (37.5%) | 2 (33.3%) | 1 (50%) | |
| Stable/progressive disease | 2 (25%) | 1 (16.7%) | 1 (50%) | |
| Overall disease response | MRD negative CR | 12(60%) | 10 (71.4%) | 2 (33.3%) |
| Partial response | 3 (15%) | 2 (14.3%) | 1 (7.1%) | |
| Stable/progressive disease | 5 (25%) | 2 (14.3%) | 3 (50%) |
| Category | Sub-category | Total subjects treated (n = 20) | CAR naïve (n = 14) | CAR pretreated (n = 6) |
|---|---|---|---|---|
| Dose level | Dose level 1 (3 × 105 CAR T cells/kg) | 4 (20%) | 1 (7.1%) | 3 (50%) |
| Dose level 2 (1 × 106 CAR T cells/kg) | 4 (20%) | 3 (21.4%) | 1 (16.7%) | |
| Dose level 3 (3 × 106 CAR T cells/kg) | 12 (60%) | 10 (71.4%) | 2 (33.3%) | |
| LD | Standard LD | 18 (90%) | 14 (100%) | 4 (66.7%) |
| Intensified LD | 2 (10%) | 0 | 2 (33.3%) | |
| CRS and ICANS* | No CRS | 10 (50%) | 5 (35.7%) | 5 (83.3%) |
| CRS grade 1-2 | 7 (35%) | 6 (42.9%) | 1 (16.7%) | |
| CRS grade 3-4 | 3 (15%) | 3 (21.4%) | 0 | |
| ICANS | 1 (5%) | 0 | 1 (16.7%) | |
| Treatment | Tocilizumab treatment | 6 (30%) | 5 (35.7%) | 1 (16.7%) |
| Corticosteroids treatment† | 4 (20%) | 3 (21.4%) | 1 (16.7%) | |
| Bone marrow response‡ | MRD negative CR | 16 (80%) | 12 (85.7%) | 4 (66.7%) |
| Partial response | 1 (5%) | 1 (7.1%) | 0 | |
| Stable/progressive disease | 3 (15%) | 1 (7.1%) | 2 (33.3%) | |
| Extramedullary response (non-CNS) (n = 8)§ | Complete response | 3 (37.5%) | 3 (50%) | 0 |
| Partial response | 3 (37.5%) | 2 (33.3%) | 1 (50%) | |
| Stable/progressive disease | 2 (25%) | 1 (16.7%) | 1 (50%) | |
| Overall disease response | MRD negative CR | 12(60%) | 10 (71.4%) | 2 (33.3%) |
| Partial response | 3 (15%) | 2 (14.3%) | 1 (7.1%) | |
| Stable/progressive disease | 5 (25%) | 2 (14.3%) | 3 (50%) |
N (%) unless otherwise specified.
Using American Society of Transplantation and Cellular Therapy (ASTCT) grading scales.
Steroids used in 1 patient for treatment of neurotoxicity only.
Nineteen of 20 patients had BM disease before treatment (1 patient was MRD-negative in the BM and had EM disease, and at 1-mo reassessments had a continued MRD-negative response in marrow and clearance of EM disease.
Eight patients had EM disease before treatment.
NSC ratings at day +14 identified 10 additional symptoms in 7 patients related to CAR T-cell therapy that were solely identified by caregivers and included new symptoms of depressed mood and distress (supplemental Table 3).
Responses
Twelve patients (60%) had a complete CR (responders = full eradication of all disease sites; Figure 1A). Sixteen (80%) achieved MRDnegative BM CR, with discrepant responses in 4 patients, all with residual or progressive EMD (supplemental Table 4). Nonresponders included 3 with PR and 5 with stable/progressive disease (Table 2). Pretreatment antigen density did not differ between responders and nonresponders (supplemental Figure 1B-C).
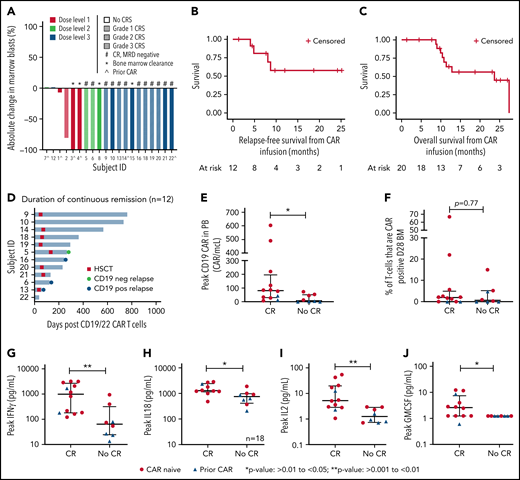
Response, outcomes, CAR expansion, and cytokine levels after CD19.22.BBζ CAR T-cell infusion. (A) Waterfall plot of the best response after CD19.22.BBζ CAR T-cell infusion. Participants were stratified by DL and CRS grade. Bone marrow clearance demonstrates those that were MRD negative by flow cytometry. (B) RFS was calculated from the date of CAR infusion until the date of relapse or last follow-up among those who went into complete remission and was not censored for HSCT. The 6- and 12-month RFS was 80.8% (95% CI: 42.4%-94.9%) and 57.7% (95% CI: 22.1%-81.9%), respectively. (C) OS was calculated from the date of CAR infusion until the date of death or last date of follow-up. The 6- and 12-month OS was 100% and 63.0% (95% CI: 35.4%-81.4%) respectively. (D) Duration of continuous remission among patients who achieved a CR. Shown are duration of remission, time to transplant if applicable, and time of relapse denoted as either antigen negative or antigen positive. (E) Peak CAR T-cell expansion in the PB assessed as absolute CAR T cells based on the percent of absolute lymphocyte count that was CAR T cell positive and stratified by CR vs no CR, and patients who were CAR naïve and CAR pretreated are denoted. (F) Peak percentage of CAR T cells in the bone marrow at day 28 assessment stratified by CR vs no CR, and patients who were CAR naïve and CAR pretreated are denoted. (G-J) Comparison of peak cytokine values of IFNγ, IL18, IL2, and GM-CSF between CR and no CR, with CAR naïve denoted with a red circle and CAR pretreated denoted with a blue triangle.
Response, outcomes, CAR expansion, and cytokine levels after CD19.22.BBζ CAR T-cell infusion. (A) Waterfall plot of the best response after CD19.22.BBζ CAR T-cell infusion. Participants were stratified by DL and CRS grade. Bone marrow clearance demonstrates those that were MRD negative by flow cytometry. (B) RFS was calculated from the date of CAR infusion until the date of relapse or last follow-up among those who went into complete remission and was not censored for HSCT. The 6- and 12-month RFS was 80.8% (95% CI: 42.4%-94.9%) and 57.7% (95% CI: 22.1%-81.9%), respectively. (C) OS was calculated from the date of CAR infusion until the date of death or last date of follow-up. The 6- and 12-month OS was 100% and 63.0% (95% CI: 35.4%-81.4%) respectively. (D) Duration of continuous remission among patients who achieved a CR. Shown are duration of remission, time to transplant if applicable, and time of relapse denoted as either antigen negative or antigen positive. (E) Peak CAR T-cell expansion in the PB assessed as absolute CAR T cells based on the percent of absolute lymphocyte count that was CAR T cell positive and stratified by CR vs no CR, and patients who were CAR naïve and CAR pretreated are denoted. (F) Peak percentage of CAR T cells in the bone marrow at day 28 assessment stratified by CR vs no CR, and patients who were CAR naïve and CAR pretreated are denoted. (G-J) Comparison of peak cytokine values of IFNγ, IL18, IL2, and GM-CSF between CR and no CR, with CAR naïve denoted with a red circle and CAR pretreated denoted with a blue triangle.
Responses were dose dependent, with no CR at DL1. A maximum tolerated dose was not achieved, and DL3 (3 × 106 CAR T cells/kg) was deemed the recommended phase 2 dose. Disease burden was not associated with response.
Comparison of CR rates between CAR-naïve and CAR-pretreated cohorts revealed important differences, albeit with small numbers. Specifically, 10 of 14 (71.4%) patients who were CAR naïve vs 2 of 6 (33.3%) patients who were CAR pretreated achieved a CR (P = .16). Notably, 3 of 4 patients treated at DL1 were CAR pretreated, and 50% of all CAR-pretreated patients were treated at DL1, limiting ability to assess if DL1 was ineffective because of CAR pretreatment or lower dose. Furthermore, both CAR pretreatment responders were treated at DL3 and received intensified LD, again precluding our ability to ascribe enhanced response to dose escalation or LD intensification. Intensified LD was, however, used for all reinfusions with 3 of 4 patients achieving a CR, including 2 with suboptimal response to the initial infusion (supplemental Appendix).
Long-term follow-up
Eight of 12 achieving CR proceeded to a consolidative HSCT at a median of 55 days (range: 46-127 days) after infusion, representing 6 first HSCTs. One patient is awaiting second HSCT. Reasons for not pursuing HSCT (n = 3) included patient preference, lack of insurance, and limited donor options for second HSCT.
With a median potential follow-up of 23.5 months (range: 1-41.4 months), the median OS was 23.7 months (95% confidence interval [CI]: 10.4-27.4 months); the median RFS was not reached (Figures 1B-C). No differences were seen in OS or RFS among CAR-naïve vs CAR-pretreated cohorts (supplemental Figure 1D-E). Eight of 12 (66.7%) remain in an ongoing remission, with a median remission duration of 256 days (range: 38-765 days; Figure 1D). Among these 8 patients, 1 remains in a durable remission > 2 years after infusion without interim therapy or consolidative HSCT; 6 proceeded to consolidative HSCT and remain in remission, and 1 is in CR awaiting HSCT.
Among the 4 who relapsed, the median time to relapse was 184 days (range: 60-265 days): 3 with CD19+ disease and 2 following consolidative HSCT. One patient who relapsed with CD19neg disease received pre-enrollment blinatumomab and no consolidative HSCT.
Clinical correlative studies
Neurocognitive testing
Seventeen of 20 (85%) completed pre- and postinfusion neurocognitive assessments. Verbal fluency testing was restricted to English-speaking participants (n = 13). At baseline, all mean Cogstate and traditional neurocognitive test scores were within normal limits (1 standard deviation from the mean), and no changes were seen in the mean standard scores after infusion across all 5 Cogstate assessments and traditional testing methods (supplemental Table 5). Importantly, most neurocognitive scores were stable or improved after infusion (supplemental Table 6).
CAR T-cell expansion
CAR T-cell expansion was seen in 18 patients with peak peripheral blood (PB) expansion at day 8. Expansion was higher in those achieving a CR than in nonresponders (median peak,. 80.4 CAR/mcL [range: 6.2-603 CAR/mcL] vs 22.6 CAR/mcL [range: 0-84.2 CAR/mcL], respectively; P = .015; Figure 1E). In BM at day 28, there were no differences between responders vs nonresponders in the percentage of T cells that were CAR+ (P = .77; Figure 1F). Expansion in both PB and day 28 BM was higher in patients who were CAR naïve compared with CAR pretreated (median PB peak, 74.59 CAR/mcL [range: 6.16-603 CAR/mcL] vs 2.6 CAR/mcL [range: 0-44.17 CAR/mcL], P = .001, and median BM peak 2.1% T cells that were CAR+ [range: 0%-67%] vs 0 [range: 0%-4.8%], respectively; P = .022).
Clinical correlative studies
Serial antigen profiling on a subset of patients demonstrated variability of antigen expression over time (supplemental Figure 1F-G). The presence of HAMA neither correlated with nor predicted response (supplemental Figure 1H-I). Intensified LD was associated with lower absolute lymphocyte count between days −2 and +3 but did not impact CAR T-cell expansion (supplemental Figures 2A-B).
Cytokine profiling revealed substantial elevations in peak secretion of interferon γ (IFNγ), IL18, IL2, IL13, and granulocyte-macrophage colony-stimulating factor (GM-CSF) in responders compared with nonresponders (P < .05; Figure 1G-J; all other cytokines, supplemental Figure 3).
CD19, CD22, and CD19/22 comparison studies
Comparison of responders across our 3 trials demonstrated that responders receiving CD22.BBζ had substantially higher peak expansion in PB and D28 BM compared with responders receiving either CD19.28ζ or CD19.22.BBζ (Figure 2A-B). Additionally, there was increased persistence of CD22.BBζ T cells compared with either CD19.28ζ or CD19.22.BBζ T cells (median: 88, 27, and 28 days, respectively; Figure 2C). Similarly, peak CRP and ferritin, alongside select cytokines, were higher in those receiving CD22.BBζ CAR T cells (Figure 2D-S).
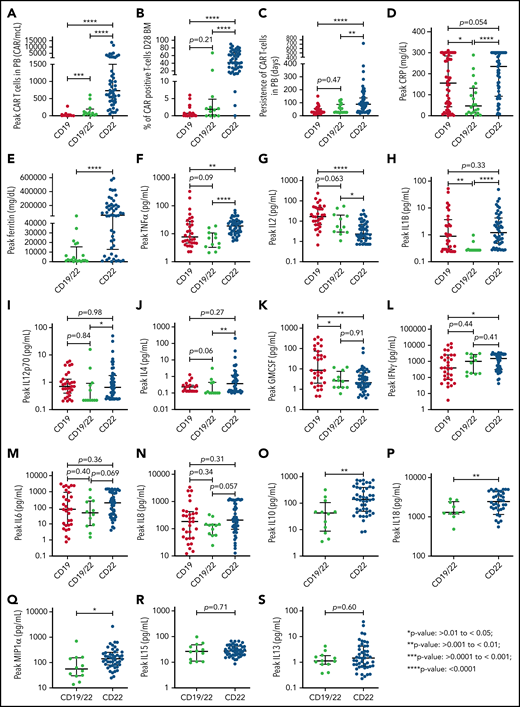
Comparison of CAR T-cell expansion, persistence, inflammatory markers, and cytokine values across 3 Pediatric Oncology Branch clinical trials. (A-B) Peak CAR T-cell expansion in the PB displayed as absolute CAR T cells and peak percentage of CAR T cells in the bone marrow, respectively, evaluated in responders across the 3 trials. (C) Persistence of CAR T cells in the PB as measured by flow cytometry. (D-E) Comparison of peak CRP and ferritin values across trials. (F-N) Comparison of peak serum cytokine values of tumor necrosis factor α, IL2, IL1B, IL4, IL12p70, IFNγ, IL6, IL8, and GM-CSF across all 3 trials. (O-S) Comparison of peak serum cytokine values of IL10, IL18, macrophage inflammatory protein-1 (MIP1)α, IL15, and IL13 between CD19/22 and CD22 CAR patients. *P > .01 to < .05; **P > .001 to < .01; ***P > .0001 to < .001; ****P < .0001.
Comparison of CAR T-cell expansion, persistence, inflammatory markers, and cytokine values across 3 Pediatric Oncology Branch clinical trials. (A-B) Peak CAR T-cell expansion in the PB displayed as absolute CAR T cells and peak percentage of CAR T cells in the bone marrow, respectively, evaluated in responders across the 3 trials. (C) Persistence of CAR T cells in the PB as measured by flow cytometry. (D-E) Comparison of peak CRP and ferritin values across trials. (F-N) Comparison of peak serum cytokine values of tumor necrosis factor α, IL2, IL1B, IL4, IL12p70, IFNγ, IL6, IL8, and GM-CSF across all 3 trials. (O-S) Comparison of peak serum cytokine values of IL10, IL18, macrophage inflammatory protein-1 (MIP1)α, IL15, and IL13 between CD19/22 and CD22 CAR patients. *P > .01 to < .05; **P > .001 to < .01; ***P > .0001 to < .001; ****P < .0001.
Reverse translational investigations
Comparison of CD19.22.BBζ CARs expressed downstream of MSCV and EF1α promoters
Given the equivalent structure of the hinge, transmembrane, and costimulatory domains across both vectors, the limited expansion and shorter persistence of CD19.22.BBζ CAR T cells compared with CD22.BBζ CAR T cells was unexpected. As the clinically generated CD19.22.BBζ CAR was expressed downstream of the retroviral MSCV promoter and CD22.BBζ was expressed downstream from the EF1α promoter, and previous studies have shown that CAR expression/function can be altered by the promoter,32,33 we compared MSCV-CD19.22.BBζ to EF1α-CD19.22.BBζ using identical manufacturing for vector and CAR T cells.
Although T-cell transduction across a large range of vector doses (2.5e6-2.5e9 ng/mL P24) was substantially higher for MSCV-CD19.22.BBζ than EF1α-CD19.22.BBζ constructs (P < .01; supplemental Figure 4A), the relative transduction of CD4 and CD8 T cells was equivalent (supplemental Figure 4B). Notably, IFNγ and IL2 secretion by MSCV-CD19.22.BBζ and EF1α-CD19.22.BBζ in response to CD19+ NALM6 ALL line over the course of 72 hours was equivalent (Figure 3A). Moreover, both MSCV-CD19.22.BBζ and EF1α-CD19.22.BBζ CAR T cells were able to eradicate CD19+CD22+ and CD19+CD22neg ALL engrafted in NSG mice during the 6-week experiment (nonsignificant; Figure 3B). This was associated with an absence of GFP+ NALM6 leukemic cells in either BM or spleen at day 42 (supplemental Figure 4C and data not shown), a similar percentage of human CD3+ T cells, and cell surface detection of both CD19- and CD22-scFv moieties (supplemental Figure 4C-D). Although cell surface detection of CD19- and CD22-scFv moieties trended toward a lower level in mice treated with MSCV-CD19.22.BBζ CAR T cells, differences were not significant (supplemental Figure 4D). Importantly, a high percentage of CAR-expressing CD8+ T cells exhibited an exhaustion phenotype at day 42, as monitored by PD1 and Tim3 expression, and this phenotype was detected irrespective of whether the CAR was expressed under the control of the MSCV or EF1α promoter (supplemental Figure 5A-B).

Efficacy of MSCV-CD19.22.BBζ CAR and EF1α-CD19.22.BBζ CAR constructs. (A) Cytokine production by EF1α-CD19.22.BBζ and MSCV-CD19.22.BBζ CAR T cells was evaluated on coculture with the indicated CD19+CD22+, CD19+CD22−, CD19−CD22+, and CD19−CD22− NALM6 leukemia lines. Cocultures were performed at a 1:1 effector/target ratio, and cytokines were monitored at 1-, 3-, 6-, 12-, 18-, 24-, 30-, 36-, 42-, 48-, 60-, and 72-hour time points on a TECAN EVO 100 robotic system. Data are representative of CAR T cells from 1 of 3 individual donors. (B) Luciferase-transduced NALM6 cells (1e6) were injected IV via tail vein into NSG mice on day 0. CAR T cells were injected at day 3, and leukemia growth was evaluated at the indicated time points by bioluminescent imaging. Quantification of bioluminescence at each time point is shown for each individual mouse (bottom graphs, P > .05). Cell surface CD19 CAR expression was evaluated using either a phycoerythrin (PE)-labeled monoclonal anti-FMC63 scFv antibody (Acro) or allophycocyanin (APC)-labeled monoclonal anti-FMC63 scFv antibody.44 CD22 CAR expression was monitored by staining with a recombinant human siglec-2/CD22 Fc chimera protein (R&D) followed by incubation with a PE- or APC-conjugated goat–anti-human immunoglobulin G (Jackson ImmunoResearch).
Efficacy of MSCV-CD19.22.BBζ CAR and EF1α-CD19.22.BBζ CAR constructs. (A) Cytokine production by EF1α-CD19.22.BBζ and MSCV-CD19.22.BBζ CAR T cells was evaluated on coculture with the indicated CD19+CD22+, CD19+CD22−, CD19−CD22+, and CD19−CD22− NALM6 leukemia lines. Cocultures were performed at a 1:1 effector/target ratio, and cytokines were monitored at 1-, 3-, 6-, 12-, 18-, 24-, 30-, 36-, 42-, 48-, 60-, and 72-hour time points on a TECAN EVO 100 robotic system. Data are representative of CAR T cells from 1 of 3 individual donors. (B) Luciferase-transduced NALM6 cells (1e6) were injected IV via tail vein into NSG mice on day 0. CAR T cells were injected at day 3, and leukemia growth was evaluated at the indicated time points by bioluminescent imaging. Quantification of bioluminescence at each time point is shown for each individual mouse (bottom graphs, P > .05). Cell surface CD19 CAR expression was evaluated using either a phycoerythrin (PE)-labeled monoclonal anti-FMC63 scFv antibody (Acro) or allophycocyanin (APC)-labeled monoclonal anti-FMC63 scFv antibody.44 CD22 CAR expression was monitored by staining with a recombinant human siglec-2/CD22 Fc chimera protein (R&D) followed by incubation with a PE- or APC-conjugated goat–anti-human immunoglobulin G (Jackson ImmunoResearch).
These experiments also revealed an attenuated response of both MSCV-CD19.22.BBζ and EF1α-CD19.22.BBζ CAR T cells to CD22 antigen.12 Neither secretion of IFNγ nor IL2 was substantially augmented in response to CD19negCD22+ NALM6 compared with an antigen-negative (CD19negCD22neg) NALM6 clone. Furthermore, CD19neg ALL continued to grow in NSG mice following adoptive transfer of CD19.22.BBζ CAR T cells (Figure 3B). This suggests that CD22 scFv activity was suboptimal in the bivalent CAR, potentially explaining the lower CD19.22.BBζ expansion seen on this study in comparison with CD22.BBζ CAR T cells. However, with some late responses detected following transfer of EF1α-CD19.22.BBζ CAR T cells (Figure 3B), we hypothesized that clinical efficacy might be improved by both moving to a bicistronic arrangement and taking advantage of the EF1α promoter.
Development of a novel CD19-22 bicistronic CAR
Hypothesizing that simultaneous and independent cell surface expression of a CD19- and CD22-scFv would overcome limitations in CD22 targeting seen with bivalent CD19.22.BBζ CAR T cells (Figure 4A), we generated 4 novel bicistronic CARs harboring CD19 and CD22 scFvs under the control of the EF1α promoter and varied CD28 and 4-1BB costimulatory domains as shown in Figure 4A ("Methods"). All CARs, irrespective of costimulatory domain, were detected at high levels at the cell surface, with linked expression (supplemental Figure S6).
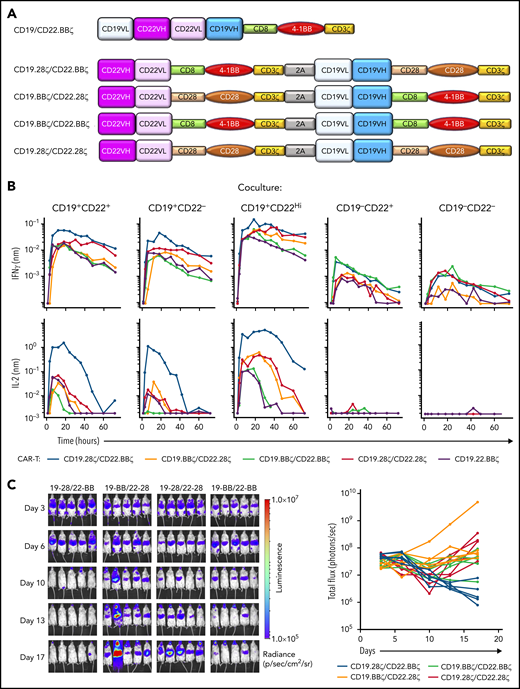
In vitro and in vivo efficacy of CD19-CD22 bicistronic CAR constructs harboring CD28 and 4-1BB costimulatory domains. (A) Schematic representation of the initial bivalent CD19.22.BBζ CAR construct and the newly generated bicistronic constructs, harboring the m971 human anti-CD22 scFv and murine FMC63 anti-CD19 scFv under the control of the EF1α promoter. Constructs differ in the CD28 and 4-1BB costimulatory domains with all hinge-transmembrane domains derived from CD28 in the former and CD8 in the latter. (B) Cytokine production induced by coculture of T cells harboring 1 of the 4 bicistronic CARs or the bivalent CD19.22.BBζ CAR (purple) was evaluated by coculture with CD19+CD22+, CD19+CD22−, CD19+CD22High, CD19−CD22+, and CD19−CD22− NALM6 lines. Cocultures were performed at a 1:1 effector/target ratio and cytokines monitored at 1-, 3-, 6-, 12-, 18-, 24-, 30-, 36-, 42-, 48-, 60-, and 72-hour time points on a TECAN EVO 100 robotic system. Results are representative of data obtained in 3 individual T-cell donors. (C) Luciferase-transduced NALM6 cells (1e6) were injected IV into NSG mice on day 0, and the indicated bicistronic CAR T cells were injected at day 3. Leukemia growth was evaluated at the indicated time points by bioluminescent imaging. Quantification of bioluminescence at each time point is shown for each individual mouse (bottom graphs).
In vitro and in vivo efficacy of CD19-CD22 bicistronic CAR constructs harboring CD28 and 4-1BB costimulatory domains. (A) Schematic representation of the initial bivalent CD19.22.BBζ CAR construct and the newly generated bicistronic constructs, harboring the m971 human anti-CD22 scFv and murine FMC63 anti-CD19 scFv under the control of the EF1α promoter. Constructs differ in the CD28 and 4-1BB costimulatory domains with all hinge-transmembrane domains derived from CD28 in the former and CD8 in the latter. (B) Cytokine production induced by coculture of T cells harboring 1 of the 4 bicistronic CARs or the bivalent CD19.22.BBζ CAR (purple) was evaluated by coculture with CD19+CD22+, CD19+CD22−, CD19+CD22High, CD19−CD22+, and CD19−CD22− NALM6 lines. Cocultures were performed at a 1:1 effector/target ratio and cytokines monitored at 1-, 3-, 6-, 12-, 18-, 24-, 30-, 36-, 42-, 48-, 60-, and 72-hour time points on a TECAN EVO 100 robotic system. Results are representative of data obtained in 3 individual T-cell donors. (C) Luciferase-transduced NALM6 cells (1e6) were injected IV into NSG mice on day 0, and the indicated bicistronic CAR T cells were injected at day 3. Leukemia growth was evaluated at the indicated time points by bioluminescent imaging. Quantification of bioluminescence at each time point is shown for each individual mouse (bottom graphs).
In assessing functionality of these CARs, ex vivo cytotoxicity studies demonstrated killing of both CD19+ and CD22+ leukemias, equivalent to that detected with single-antigen targeted CARs (supplemental Figure 6B). Furthermore, bicistronic CAR T cells exhibited increased killing of CD19neg/CD22+ leukemia compared with T cells harboring the bivalent EF1α-CD19.22.BBζ, at both 1:1 and 1:5 effector/target ratios (supplemental Figure 6B). Moreover, high-throughput robotics analyses of these bicistronic constructs, evaluating IFNγ and IL2 secretion in response to NALM6 leukemic cells with different levels of CD19 and CD22 expression, revealed differential activity. Briefly, compared with EF1α-CD19.22.BBζ CAR T cells, the EF1α-CD19.28ζ/CD22.BBζ CAR T-cell construct exhibited much higher levels of IL2 secretion against CD19+ and CD22+ leukemias (Figure 4B). Moreover, cytokine secretion of EF1α-CD19.28ζ/CD22.BBζ CAR T cells against a NALM6 line engineered to express higher levels of CD22 (CD22High) was increased compared with the parental line (Figure 4B). Notably, these bicistronic constructs did not exhibit nonspecific cytokine secretion, as only minimal cytokines were detected in response to CD19negCD22neg NALM6. Overall, these data correlated with a robust killing of CD19+CD22+ NALM6 leukemia by CD19.28ζ/CD22.BBζ CAR T cells in NSG mice at levels higher than that detected with alternative constructs tested (Figure 4C) and support clinical translation of this novel EF1α-CD19.28ζ/CD22.BBζ CAR T-cell construct.
Discussion
Achieving a sustainable remission after CAR T-cell therapy in B-ALL is dually constrained by limitations in functional CAR T-cell persistence and leukemic antigen escape. Building on our institutional experiences with CD19.28ζ4,5 and CD22.BBζ CAR T cells,10,11 we developed and translated a novel bivalent CD19.22.BBζ with the hypothesis that a dual-antigen targeted CAR T cell could overcome the limitations of single-antigen targeting.12 Importantly, we demonstrate clear efficacy in a heavily pretreated population with a low toxicity profile, with only 1 patient experiencing ICANS and 3 patients achieving CR without CRS. Although our results demonstrate safety, feasibility, and efficacy, a host of limitations emerged, prompting additional investigations.
By virtue of our institutional iterative experience, head-to-head comparisons across 3 constructs revealed that peak expansion and persistence of CD19.22.BBζ CAR T cells resembled our CD19.28ζ CAR but differed substantially from CD22.BBζ CAR. Although both CD19.22.BBζ and CD22.BBζ CAR are lentiviral-based constructs, the former was generated with the MSCV promoter and the latter with EF1α. Of note, our CD19/28ζ CAR incorporated MSCV on a retroviral backbone.34 Given the importance of the promoter in directing transgene expression and thereby CAR T-cell function,32,33,35 our reverse translational studies focused on preclinical exploration of MSCV vs EF1α to understand the impact of the promoter on function. Although our laboratory investigations demonstrated no major difference in CAR T-cell efficacy between the 2 promoters, the observation that a significant percentage of these CARs exhibited a phenotypic signature of exhaustion, as monitored by the expression of PD1 and Tim3, warrants further study.
We hypothesize that suboptimal targeting of CD22 by the bivalent CD19.22.BBζ CAR used by both our group and Stanford36 may account for its differential expansion and persistence. Differences across manufacturing methodologies, platform, and vector design likely also contributed to differences across the 3 trials. Nonetheless, with the goal of improving dual targeting functionality, we developed novel bicistronic constructs to facilitate independent expression of CD19 and CD22 CARs. Notably, we found that an EF1α-promoted CD19.28ζ/CD22.BBζ CAR has improved ability to target CD22. With improved CD22 targeting, we hypothesize that persistence and expansion may more closely resemble the experience with single-antigen targeted CD22.BBζ CAR T cells. Accordingly, this construct is currently being tested in adolescents and adults with NHL (#NCT05098613) with a clinical trial in CAYA B-ALL forthcoming.
Interestingly, persistence of other dually targeted CARs incorporating 4-1BB, including co-administration18 and co-transduction models,20,37 has been similarly limited, with rare exceptions.38,39 Whether transduction of more complex vectors impact T-cell phenotype, expansion, and/or persistence warrants close monitoring, particularly with the rapid progress in multiantigen targeting strategies. Future analyses to explore the activation/exhaustion immunophenotype of the infused product in relationship to function are planned.
In the present context of multiple US Food and Drug Administration–approved CD19 CAR T-cell constructs for B-ALL/NHL and improved availability of investigational CD19- and CD22-targeted CAR T-cell constructs, it will be critical to determine how prior CAR T-cell therapy impacts patient responsiveness to novel CAR T-cell constructs, especially when the same antigen is being targeted. Importantly, responsiveness of patients to a second CAR infusion targeting a unique antigen (eg, CD22) has not been impeded by prior CD19 CAR.10 However, the development of immunogenicity against CAR T cells can be problematic, for example, with repeated infusions of CAR T cells incorporating FMC63, a murine-based CD19 scFv.7,11,40-42 In the present study, HAMA analysis was not able to predict between responders/nonresponders or discriminate between patients who were CAR pretreated, potentially because HAMA may be insufficient for identifying immunogenicity related to prior CAR T-cell exposure.42
Given limited efficacy of second infusions43 and in patients who were CAR pretreated in this study, considering prior CAR T-cell exposure when testing novel constructs will be critical. Importantly, however, CAR pretreatment did not preclude response, and LD intensification may serve to improve the likelihood of response, supported by our limited experience with reinfusion. Further experience with intensified LD is needed to assess if this approach can improve outcomes and inform study design for future CAR T-cell trials, particularly important with increasing cases of post-CAR relapse. Patients with EMD remain another challenging cohort, exemplified by our patients with discrepant responses. Our recent cross-trial analysis further highlights challenges of EMD treatment, particularly in those with multifocal disease.24
As this new bicistronic construct is implemented, our current CD19.22.BBζ trial will shift toward treatment of CNS3 disease given both limited ICANS and favorable neurocognitive testing results, which supports extending the therapeutic index of CD19.22.BBζ CAR T-cells. The prospective incorporation of neurocognitive testing, a particularly salient feature of our protocols, is imperative, particularly in younger patients and in those with CNS involvement. The pilot use of Cogstate, a computerized test battery of neurocognitive assessments with alternate test forms to reduce practice effect, was determined to be easy to administer and feasible to use in this patient population, supporting its use in future CAR T-cell trials. Importantly, as Cogstate is available in multiple languages, increasing accessibility of neurotoxicity assessment tools, particularly for those whom English is not a first language, is a critical next step to improve applicability.
In conclusion, this novel bivalent CD19.22.BBζ CAR T cell was well tolerated and highly effective in CAYA with B-ALL, including in those who were CAR pretreated. However, the truncated persistence and suboptimal CD22 targeting were clear limitations, and improving on dual functionality and persistence is needed to prevent antigen escape and maintain remission. With the goal of facilitating the independent targeting of CD19 and CD22, our iterative design of bicistronic constructs has identified the EF1α-promoted expression of CD19.28ζ and CD22.BBζ CARs as the next combinatorial strategy to be taken into the clinic.
Acknowledgments
The authors gratefully acknowledge the study participants and their families, referring medical care teams, the faculty and staff of the National Institutes of Health (NIH) Clinical Center who provide their expertise in the management of the study participants, patient care coordinators, and the data managers, Showri Kakumanu and Ekaterina Nikitina, involved with this work. The authors additionally acknowledge Melissa Baker Nour Al Griwhati and Amanda Rhodes for support of the neurocognitive assessments and/or data management on this study.
This work was supported in part by the Center for Cancer Research, Intramural Research Program, National Cancer Institute (grant ZIA BC 011823 to N.N.S. and grant ZIA BC 011923 to N.T.), the NIH Intramural Research Program through a National Cancer Institute FLEX award, NIH Clinical Center, NIH (to G.A.-B., N.T., and N.N.S.). Additionally, this project has been funded in whole or in part with federal funds from the National Cancer Institute, NIH, under contract 75N91019D00024 (to M.A.T.-T.). Research funding was also provided by the Children’s Cancer Foundation (to H.S.)
The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
Authorship
Contribution: H.S. and N.N.S. designed the study, performed primary data analysis, led the clinical trial, and wrote the first version of the manuscript; N.T. provided oversight of preclinical studies, contributed to writing the first version of the manuscript, and performed primary data analysis; H.Q. and A.S. designed the preclinical studies, performed data analysis, and wrote select sections; T.J.F., J.A.L., S.S., H.-W.W., C.M.Y., P.L.W., and M.A. provided critical input and contributed to writing of selection sections within the manuscript; P.L.W. and S.M. designed and performed neurocognitive testing, and P.L.W. conducted the analyses of the neurocognitive and NSC data; M.A.T.-T. performed neurocognitive testing and data management and contributed to writing select sections of the supplement. S.M.S. conducted statistical analysis and provided critical input on select sections within the manuscript; S.L.H., S.P., and D.S. provided support for all elements of cell manufacturing; H.S., N.N.S., J.A.L., S.S., B.Y., S.A.S., L.L., T.F., and J.C.M. provided patient care and collected and analyzed patient data; K.D., M.B., S.G., S.A., C.D.C., D.S., M.P., and G.A.-B. conducted preclinical studies and analyzed relevant data; Y.W. and J.I. performed cytokine profiling and HAMA assays; D.W.L. and C.L.M. provided data related to the CD19/28ζ CAR T-cell trial; No non-author wrote the first draft or any part of the paper; and all authors contributed to the review of the final manuscript and have agreed to be co-authors.
Conflict-of-interest disclosure: D.W.L. consults for Harpoon Therapeutics, serves on external advisory boards for BMS/Juno Therapeutics and Amgen, and his institution (UVA) receives clinical trial support from Gilead/Kite Pharma. C.L.M. consults for Lyell, Syncopation, Nektar, Bristol Myers Squibb, Glaxo Smith Kline, Immatics, Neoimmune Tech, Apricity, Mammoth, and Ensoma; holds equity in Lyell, Syncopation, Apricity, Mammoth, and Ensoma; and receives research funding from Lyell. D.S. is an employee of Lentigen, a Miltenyi Biotec Company. H.Q. and T.J.F. are co-inventors of the bivalent and bicistronic CAR constructs. H.Q. and T.J.F. have filed US patent number 10738 116: Dual specific anti-CD22-anti-CD19 Chimeric Antigen Receptors. T.J.F. is an employee of Sana Biotechnology. The remaining authors declare no competing financial interests.
Correspondence: Haneen Shalabi, Pediatric Oncology Branch, National Cancer Institute, National Institutes of Health, Building 10, Room 1W-5750, 9000 Rockville Pike, Bethesda, MD 20892-1104; e-mail: haneen.shalabi@nih.gov; Naomi Taylor, Pediatric Oncology Branch, National Cancer Institute, National Institutes of Health, Building 10, Room 1W-3750, 9000 Rockville Pike, Bethesda, MD 20892-1104; e-mail: taylorn4@mail.nih.gov; and Nirali N. Shah, Pediatric Oncology Branch, National Cancer Institute, National Institutes of Health, Building 10, Room 1W-5750, 9000 Rockville Pike, Bethesda, MD 20892-1104; e-mail: nirali.shah@nih.gov.
Data that support the findings of this study are available from the corresponding authors upon reasonable request.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
