

In this issue of Blood, Shalabi et al1 report on the safety, efficacy, and clinical limitations of a novel bivalent MSCV-CD19/CD22-4-1BB (CD19.22.BBζ) chimeric antigen receptor (CAR) T-cell in heavily pretreated patients with B-cell acute lymphoblastic leukemia, including those previously treated with CD19- or CD22-directed CAR T cells.
This report is timely, given the high remission rates after treatment with CAR T cells that target dual B-cell antigens, which has enabled consolidative stem cell transplantation and disease-free survival in some patients.2-5 Furthermore, as the use of (and indications for) commercial and investigational CAR T cells targeting CD19 or CD22 increases, the number of patients seeking treatment with a dual-targeted CAR T-cell product after previous CAR T-cell therapy has also increased. Treating patients who previously received CAR T cells with a subsequent autologous CAR T-cell therapy broaches new unanswered questions, which the authors begin to address in their article.
In their phase 1 trial, Shalabi et al found that CD19.22.BBζ CAR T cells were well-tolerated and effective, with 12 of 20 patients achieving complete responses and an additional 4 patients clearing marrow disease, albeit with residual extramedullary disease. Compared with patients in similar trials, a larger proportion of patients in their trial (40%) had evidence of extramedullary disease at enrollment. This highlights the refractory nature of the disease and raises questions regarding the trafficking of CAR T cells to extramedullary sites such as the central nervous system along with efficacy at these sanctuary sites.6 In addition, 30% of patients in their study had received previous CAR T-cell therapy.
Multi-antigen–targeting CARs are believed to reduce antigen modulation and thus offset the risk of antigen escape, a phenomenon observed after therapy with CARs that target a single antigen.7,8 However, the best strategy for developing dual-targeting CARs has not yet been established. The use of a bivalent vector (tandem or bicistronic) allows for uniform expression of both targeting moieties (single chain variable fragments [scFv’s]) and the relative ease of manufacturing compared with co-transduction or co-infusion.9 Designing a bivalent CAR requires complex vectors consisting of variable regions that influence the expansion, function, and persistence of the final product. Furthermore, the promoter region of the CAR vector has an impact on T-cell properties, including transgene expression, transduction, surface density, and function.10
Shalabi et al identified some limitations of their construct after infusion, noting decreased expansion and persistence compared with the previously studied human elongation factor 1α (EF1α)-CD22.BBζ CAR. In a series of experiments, they further probed the mechanisms responsible for these observations. The authors hypothesized that the decreased expansion and persistence of their murine stem cell virus (MSCV) bivalent CAR was a result of the difference in promoter region because the CAR was otherwise comparable to their EF1α-CD22.BBζ CAR. In an NALM-6 NSG (NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ [The Jackson Laboratory]) mouse model, the authors found that CD19.22BBζ CAR–expressing CD8+ T cells incorporating either the MSCV or EF1α promoter region demonstrated an exhausted phenotype. Although pretreatment antigen density did not correlate with response (as in previous reports regarding CD19/CD22 CAR T cells2,3), CD19.22BBζ CAR T cells containing either promoter had attenuated responses to CD22 compared with CD22.BBζ CAR T cells. Previous studies reported that diminished responses to CD22 can limit in vivo expansion and functionality of other dual-targeting products,2 which underscores the importance of efforts to enhance CD22 targeting in dual-targeting CAR T cells. Suboptimal CD22 scFv activity seems to limit the functionality of the CD19 component as well.
Limited persistence has been reported in other clinical trials of dual-targeted CD19/CD22 CAR T cells,2-5 and a variety of factors influence attenuated expansion and persistence. These factors include the design of the CAR, limited binding of the CAR with its cognate antigen (as reported by Shalabi et al), reduced CAR signaling, and exhausted T-cell phenotype correlating with abrogated function.11 In the AUTO3 study (United Kingdom), the authors found that enhanced persistence was associated with a higher proportion of CD4- or CD8-naïve cells within the CAR product, regardless of treatment dose, and low programmed cell death protein 1 (PD1) expression at peak expansion.2
In the setting of reduced persistence, especially in patients who received a previous CAR T-cell product, immunological rejection of CAR T cells must be considered because scFv’s are often derived from mice. Although there was no apparent correlation between response and the presence of human anti-mouse antibodies (HAMAs) in the Shalabi et al study, measurements of HAMAs were limited to the first month. Assessment of HAMAs and evidence of T-cell–mediated rejection at later timepoints will be key in dissecting the impact of each of these factors on CAR T-cell function. Future studies should interrogate these factors (which will vary by construct and disease type) in a reverse translational manner to understand clinical observations and improve CAR T-cell products.
To troubleshoot the attenuated response to CD22, Shalabi et al tested 4 bicistronic (as opposed to tandem) CD19/CD22 CAR vectors containing the EF1α promoter with various co-stimulatory domains. EF1α-CD19.28ζ/CD22.BBζ CAR T cells demonstrated increased cytokine secretion and cytotoxicity against CD19/CD22-expressing cell lines, particularly lines engineered to have higher CD22 expression or lacking CD19 expression, as well as enhanced NALM-6 killing in NSG mice.
In summary, Shalabi et al demonstrated that dual-targeting CD19/CD22 CAR T cells are safe with promising clinical results and that suboptimal CD22 targeting may explain their reduced persistence. Despite the small cohort, this study highlights the importance of post-clinical exploration of these increasingly common but complex vectors that target multiple antigens. This benchwork to understand the molecular drivers of clinical observations will enable engineering of CAR T cells with the potential for prolonged persistence (see figure). Until then, the Shalabi et al study also underscores the necessity of consolidative allogeneic hematopoietic stem cell transplantation after treatment with dual-targeting CAR T cells, given their limited persistence.
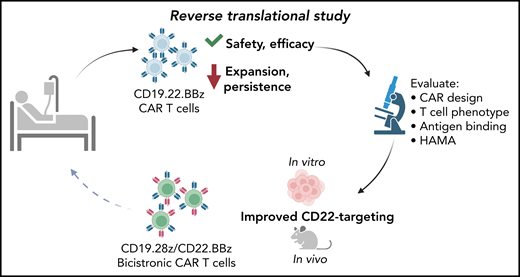
Schematic of reverse translational study as described by Shalabi et al. From left to right: participants were treated with CD19.22.BBz CAR T cells in the described clinical trial, demonstrating safety and efficacy but decreased expansion and persistence compared to prior CD22.BBz CAR T trials. Reverse translational (bedside-to-bench) studies revealed CAR design, T-cell phenotype, antigen binding capacity and HAMA-mediated rejection as potential mechanisms to explain the clinical findings. The authors concluded that improved CD22-targeting would enhance the function of dual-targeting CAR T cells, testing this hypothesis in vitro and in an in vivo mouse model. These studies led to the development of an improved dual-targeting CD19.28z/CD22.BBz bicistronic CAR that will be evaluated in participants in an upcoming clinical trial.
Schematic of reverse translational study as described by Shalabi et al. From left to right: participants were treated with CD19.22.BBz CAR T cells in the described clinical trial, demonstrating safety and efficacy but decreased expansion and persistence compared to prior CD22.BBz CAR T trials. Reverse translational (bedside-to-bench) studies revealed CAR design, T-cell phenotype, antigen binding capacity and HAMA-mediated rejection as potential mechanisms to explain the clinical findings. The authors concluded that improved CD22-targeting would enhance the function of dual-targeting CAR T cells, testing this hypothesis in vitro and in an in vivo mouse model. These studies led to the development of an improved dual-targeting CD19.28z/CD22.BBz bicistronic CAR that will be evaluated in participants in an upcoming clinical trial.
By designing an alternative CAR, the authors improved dual functionality and plan to evaluate their EF1α-CD19.28ζ/CD22.BBζ CAR T cells in future clinical trials. Given the more exhaustive phenotype seen in both MSCV and EF1α constructs, it will be important to correlate extended phenotypic data collected on infused EF1α-CD19.28ζ/CD22.BBζ CAR products with clinical expansion and persistence. This and other CD19/CD22 studies demonstrate that persistence and potency against CD22 are both critical to the success of this therapy and to the durability of remission. Future exploration will address several unanswered questions: What is the ideal phenotype of dual-targeting CAR T cells? How does previous treatment with murine CAR T cells impact immune-mediated rejection? Do humanized CAR T cells effectively mitigate this risk? And, will iterative optimization of these products prolong persistence? The answers to these questions may position dual-targeting CAR T cells as a stand-alone therapy without the need for consolidative transplantation.
Conflict-of-interest disclosure: R.H.R. serves as a consultant for Pfizer, has received honoraria from Novartis, and has received research funding from Tessa Therapeutics and Kite Pharma/Gilead. L.S. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal