

Abstract
Comparison of treatment strategies in de novo pediatric acute lymphoblastic leukemia (ALL) requires standardized measures of efficacy. Key parameters that define disease-related events, including complete remission (CR), treatment failure (TF; not achieving CR), and relapse (loss of CR) require an updated consensus incorporating modern diagnostics. We collected the definitions of CR, TF, and relapse from recent and current pediatric clinical trials for the treatment of ALL, including the key components of response evaluation (timing, anatomic sites, detection methods, and thresholds) and found significant heterogeneity, most notably in the definition of TF. Representatives of the major international ALL clinical trial groups convened to establish consensus definitions. CR should be defined at a time point no earlier than at the end of induction and should include the reduction of blasts below a specific threshold in bone marrow and extramedullary sites, incorporating minimal residual disease (MRD) techniques for marrow evaluations. TF should be defined as failure to achieve CR by a prespecified time point in therapy. Relapse can only be defined in patients who have achieved CR and must include a specific threshold of leukemic cells in the bone marrow confirmed by MRD, the detection of central nervous system leukemia, or documentation of extramedullary disease. Definitions of TF and relapse should harmonize with eligibility criteria for clinical trials in relapsed/refractory ALL. These consensus definitions will enhance the ability to compare outcomes across pediatric ALL trials and facilitate development of future international collaborative trials.
Introduction
Children with newly diagnosed acute lymphoblastic leukemia (ALL) in high-income countries are usually treated in clinical trials encompassing different therapeutic approaches. Treatment strategies and overall results are quite comparable among national and international study groups despite treatment variations.1-3 There is no uniform consensus on the basic definitions of response criteria, including complete remission (CR), treatment failure (TF, sometimes also referred to as non-CR, primary treatment failure, induction failure, or refractory disease), and relapse in pediatric ALL. These definitions are highly relevant to treatment decisions; for example, when to conclude that the frontline therapy is only partially effective (prompting stratification into a more intensive treatment arm) or largely ineffective (prompting use of alternative or salvage therapy). Up to approximately the year 2000, morphologic evaluation (cytology) was the primary method to establish the diagnosis of ALL and to assess the treatment response. Twenty-five percent or more lymphoblasts in the bone marrow (BM) defined the diagnosis of ALL and BM relapse.4,5 Since 2000, new diagnostic methods, able to detect leukemic cells with greater sensitivity and specificity compared with morphology, have emerged. Molecular and immunophenotypic techniques can measure minimal residual disease (MRD) reliably at the threshold of 1 in 10 000 to 100 000 cells. Response assessment by MRD detection has become the most important independent predictor of treatment outcome in many studies.6-8
The objective of all pediatric ALL study groups is to increase the event-free survival (EFS) and overall survival (OS) rates and improve short- and long-term quality of life. Because of the numerous new therapeutic options being investigated by various study groups worldwide, it is crucial to be able to compare results between different protocols. Thus, standardized definitions of CR and TF after the initial therapy (assessed after a comparable treatment period) and standardized definitions of relapse after achieving CR are required. Common definitions of TF and relapse may provide more uniform eligibility criteria for relapsed/refractory studies. In pediatric oncology, there are other well-established examples of uniform consensus definitions of treatment response and failure being used routinely in clinical trials (eg, response evaluation criteria in solid tumors [RECIST], solid tumors; Cheson, acute myeloid leukemia; Lugano, Hodgkin disease).9-11
A key question is which parameters in the evaluation of outcome are most relevant in upfront ALL trials. OS (time-to-death) is important, but it provides incomplete information. The single most widely accepted parameter is EFS. The main advantage of EFS over OS is the incorporation of clinically important outcomes in addition to death, such as the inability to achieve CR (TF) or relapse (both of which reflect unsatisfactory response to frontline treatment requiring salvage therapy), therapy-related mortality (induction death and death in CR, caused by resistant disease or treatment-related toxicity), and second malignancies. Furthermore, OS in a clinical trial is influenced by the efficacy of salvage therapy and thus may not inform about the efficacy of frontline therapy.
For calculation of time-to-event from diagnosis, the following parameters must be defined consistently: (1) CR (first CR; because relapse can only occur if CR has been achieved); (2) primary treatment failure (TF; not achieving CR at a predefined time point; eg, end of induction or end of consolidation); and (3) relapse (re-emergence of leukemia after CR has been achieved).
To address these issues, the Ponte-di-Legno (PDL) group representatives endeavored first to collect and summarize the current definitions of CR, TF, relapse, and other events in the pediatric ALL study groups and then to create consensus definitions to be used in future clinical trials.
Methods
This collaborative project of the PDL group comprises the following study groups with their recent protocols: Associazone Italiana Ematologia Oncologia Pediatrica (AIEOP), Berlin-Frankfurt-Münster group (BFM) (AIEOP-BFM ALL 2017); Intercontinental BFM group for ALL (ALL-IC BFM) (ALL-IC BFM 2009); ALLtogether (ALLTogether1); Co-operative Study Group (COALL) (CoALL 08-09); Children's Oncology Group (COG) (AALL1731, AALL1732, AALL1721); Dutch Childhood Oncology Group (DCOG) (DCOG-ALL-11); Dana Farber Cancer Institute (DFCI) ALL Consortium (DFCI 16-001); European Study Group for Philadelphia positive ALL (EsPhALL)/COG (EsPhALL2017/COG (AALL1631); Japan Children's Cancer Group (JCCG) (JPLSG-ALL-B12), Nordic Society of Paediatric Haematology and Oncology (NOPHO) (NOPHOALL 2008); Spanish Society of Paediatric Haematology Oncology (SEHOP Group) and Programa para el Tratamiento de Hemopatías Malignas (PETHEMA) (ALL/SEHOP-PETHEMA 2013); Société Française de lutte contre les Cancers de l'Enfant et de l'Adolescent (SFCE) (CAALL F01); St. Jude Children's Research Hospital (SJCRH) (TOT-XVI); Taiwan Pediatric Oncology Group (TPOG) (TPOG-ALL2013); and United Kingdom Acute Lymphoblastic leukaemia (UKALL) (UKALL 2011 Trial).12-27
Initially, recent protocols were reviewed for the definitions of CR, TF, relapse, and events. A summary was provided to all participant study groups to review the definitions derived from these study protocols. In preparation for a face-to-face meeting of the PDL group in Zbiroh, Czech Republic, on 19 May 2019, an overview of all definitions was prepared, and the first classifications were made.
Terminology
BM and central nervous system involvement
The following morphologic classifications of BM and central nervous system (CNS) involvement with ALL were developed decades ago and will be referred to in the presentation of the results.5,28,29
BM classification by cytomorphology is defined: (1) M1 marrow, <5% blasts; (2) M2 marrow, ≥5 to <25% blasts; (3) M3 marrow, ≥25% blasts. Of note, COG has historically defined M3 marrow as >25% blasts rather than ≥25% blasts.28
CNS classification by cytomorphology and/or clinical findings (±imaging and biopsy):30 (1) CNS1, absence of blasts on cytospin preparation in cerebrospinal fluid (CSF) and no clinical or imaging findings of CNS leukemia; (2) CNS2, ≤5/μL nucleated cells in CSF, cytospin preparation positive for blasts, and no clinical or imaging findings of CNS disease; (3) CNS3: >5/μL nucleated cells in CSF and cytospin preparation positive for blasts or clinical or imaging findings of CNS disease. Of note, COG, EsPhALL/COG, DFCI, NOPHO, SFCE, SJCRH, and TPOG defined CNS3 as ≥5/μL nucleated cells in CSF.31
Treatment phases
In most protocols, induction chemotherapy (phase Ia in BFM-type regimens) lasts about 4 to 6 weeks and is followed by a second phase, frequently known as consolidation phase (phase Ib in BFM-type regimens), then a third intensification phase often targeting the extramedullary space, and finally a fourth reintensification phase (delayed intensification) with or without CNS irradiation, depending on risk stratification and treatment strategy. In a small minority of cases, the latter parts of the protocol sequence are replaced by allogeneic hematopoietic stem cell transplantation.32
Results
Complete remission
Large differences among groups were found in definitions of CR, reflecting differences in timing of CR assessment, in threshold levels of leukemic cells to define CR, and the detection method (Table 1; supplemental Table 1, available on the Blood Web site).
Definition, methods, and time points for remission assessment in current treatment protocols for pediatric ALL (remission requires fulfillment of all sites)
| Anatomic sites | Methods | Study group | Time point, d | Exceptions |
|---|---|---|---|---|
| BM | ||||
| <5% blasts in BM | Cytomorphology | AIEOP-BFM ALL-IC BFM CoALL DCOG DFCI JCCG TPOG | 33, EOI 33, EOI 29, EOI 33, EOI 32, EOI 33, EOI 35-42, EOI | |
| <5% blasts in BM | Cytomorphology plus confirmation by FCM-/PCR-MRD/genetics | NOPHO SEHOP-PETHEMA SFCE SJCRH | 29, EOI EOI 35-42, EOI 38-42, EOI | SJCRH mainly uses FCM-MRD |
| <5% blasts in BM | PCR-MRD (±cytomorphology) | UKALL | 29, EOI | |
| <1% blasts in BM | FCM-/PCR-MRD (±cytomorphology) | ALLTogether COG EsPhALL/COG | EOI as the earliest EOI for early CR, EOC for late CR End of Cons. block 3 | |
| CNS | ||||
| CNS 1 | Cytomorphology, imaging, clinical examination | Every participating study group | See above for respective study group | |
| CNS 1 | Cytomorphology plus confirmation by FCM-MRD | ALLTogether | See above for respective study group | If suspect cells are seen; evaluation at EOC (d 71) |
| EM | ||||
| Resolution of leukemic infiltrates | Clinical examination, imaging, histology | ALL-IC COG DCOG JCCG EsPhALL/COG | See above for respective study group | DCOG: retesting of testicular involvement after Prot. M NOPHO: retesting EM involvement at d 85 |
| Reduction of initial leukemic mass to 1/3 | Clinical examination, imaging, histology | AIEOP-BFM ALLTogether CoALL DFCI SEHOP-PETHEMA SFCE UKALL | See above for respective study group | UKALL: retesting of testicular involvement at wk 8 |
| Anatomic sites | Methods | Study group | Time point, d | Exceptions |
|---|---|---|---|---|
| BM | ||||
| <5% blasts in BM | Cytomorphology | AIEOP-BFM ALL-IC BFM CoALL DCOG DFCI JCCG TPOG | 33, EOI 33, EOI 29, EOI 33, EOI 32, EOI 33, EOI 35-42, EOI | |
| <5% blasts in BM | Cytomorphology plus confirmation by FCM-/PCR-MRD/genetics | NOPHO SEHOP-PETHEMA SFCE SJCRH | 29, EOI EOI 35-42, EOI 38-42, EOI | SJCRH mainly uses FCM-MRD |
| <5% blasts in BM | PCR-MRD (±cytomorphology) | UKALL | 29, EOI | |
| <1% blasts in BM | FCM-/PCR-MRD (±cytomorphology) | ALLTogether COG EsPhALL/COG | EOI as the earliest EOI for early CR, EOC for late CR End of Cons. block 3 | |
| CNS | ||||
| CNS 1 | Cytomorphology, imaging, clinical examination | Every participating study group | See above for respective study group | |
| CNS 1 | Cytomorphology plus confirmation by FCM-MRD | ALLTogether | See above for respective study group | If suspect cells are seen; evaluation at EOC (d 71) |
| EM | ||||
| Resolution of leukemic infiltrates | Clinical examination, imaging, histology | ALL-IC COG DCOG JCCG EsPhALL/COG | See above for respective study group | DCOG: retesting of testicular involvement after Prot. M NOPHO: retesting EM involvement at d 85 |
| Reduction of initial leukemic mass to 1/3 | Clinical examination, imaging, histology | AIEOP-BFM ALLTogether CoALL DFCI SEHOP-PETHEMA SFCE UKALL | See above for respective study group | UKALL: retesting of testicular involvement at wk 8 |
BM: bone marrow, CNS: central nervous system, d: day, EM: extramedullary sites, EOC: end of consolidation, EOI: end of induction, FCM-MRD: flow cytometric minimal residual disease, MRD: minimal residual disease, PCR-MRD: polymerase chain reaction minimal residual disease.
Time point(s) of remission assessment
All study groups agreed that the earliest assessment of CR should occur no sooner than the completion of induction chemotherapy. Remission assessment at the end of induction is used for risk stratification in every study group except EsPhALL/COG. In some protocols (AIEOP-BFM ALL 2017, ALL-IC BFM 2009, ALLTogether1, AALL1731, CoALL 08-09, DCOG-ALL-11, NOPHO-ALL 2008, DFCI 16-001), remission status is also explicitly re-examined at later therapy time points for patients not in CR (eg, after high-intensity intensification blocks).
Anatomic sites included in remission assessment
All study groups evaluate CR in BM and CNS. All groups except SJCRH and TPOG also evaluate CR in other extramedullary sites.
Thresholds of lymphoblasts in BM
COG and ALLTogether currently use a threshold of <1% lymphoblasts in BM as the criterion for CR. This is a new MRD-based addition to current definitions and was not used in earlier trials. All other study groups continue to use a historical threshold of <5% lymphoblasts in BM as a criterion for CR.
Detection methods for response assessment
BM
Methods for detection of leukemic cells have been described in the study protocols, particularly for BM assessment, and as MRD techniques have evolved, the variability has increased. CR assessment is currently based solely (without incorporating MRD methodology) on cytomorphology in AIEOP-BFM, ALL-IC BFM, CoALL, DCOG, DFCI, JCCG, and TPOG, and thus, the morphologic classification of the BM in M1, M2, and M3 applies.28 In unclear cases, alternative methods are applied. NOPHO and SEHOP-PETHEMA initially assess the therapy response cytomorphologically but require confirmation of a result with <5% lymphoblasts by flow cytometry (FCM). If the results differ, FCM takes precedence.33 SFCE also examines the bone marrow cytomorphologically and then uses polymerase chain reaction-MRD (PCR-MRD) as a confirmation method. SJCRH mainly uses FCM for remission assessment. COG, ALLTogether, and UKALL consider FCM- and/or PCR-MRD as the primary source of information and only consider cytomorphological results as a backup in the unlikely situation of failure of MRD technologies. In EsPhALL/COG, CR is primarily determined by PCR-MRD, but backup by FCM-MRD is permitted to assess CR if PCR-MRD was unavailable, and lack of CR can be confirmed by cytomorphology (M2/M3) or by an additional method (PCR-MRD, FCM-MRD, fluorescence in situ hybridization [FISH]).33-36
CSF
Cytomorphology is used by all study groups to determine CR status in CSF. The presence of CNS leukemia is classified according to published criteria.29,31,37 SJCRH uses indirect nuclear terminal deoxynucleotidyl transferase immunofluorescence assay to confirm the leukemic nature of blasts in CSF. ALLTogether and CoALL recommend a differentiation of malignant and nonmalignant contamination with FCM-MRD for abnormal CSF findings.38 In addition, all groups do clinical examinations and imaging in case of neurologic symptoms. In exceptional cases where imaging is equivocal, biopsy may be required.
Other extramedullary sites
In all study groups except SJCRH and TPOG, non-CNS extramedullary (EM) sites are examined clinically by imaging, and/or biopsy to assess reduction of tumor mass or to assess a viability of residual tumor tissue histologically. ALL-IC, COG, DCOG, JCCG, and EsPhALL/COG require a resolution of EM leukemia. AIEOP-BFM, ALLTogether, CoALL, DFCI, SEHOP-PETHEMA, SFCE, and UKALL require a reduction of initial tumor mass (especially mediastinal mass) to at least one-third of the initial size. NOPHO evaluates EM leukemia after consolidation therapy for the first time. Some study groups (UKALL, DCOG, and NOPHO) do not incorporate resolution of testicular disease at the end of induction for assessment of CR; they assess testicular response at later therapy time points for the first time.
TF
Significant differences among groups were found in definitions of TF as events (Table 2; supplemental Table 1). Although there is a broad consensus that TF should be defined as failure to achieve a CR before a prespecified time point, there are significant differences in the prespecified time point.
Definitions of treatment failure events in current treatment protocols
| Event | Methods | Study group |
|---|---|---|
| Induction failure (EOI) ≥5% blasts in BM | Cytomorphology and/or FCM-/PCR-MRD or genetics | JCCG, SEHOP-PETHEMA, SFCE, SJCRH, TPOG |
| Induction failure (EOI) ≥25% blasts in BM | See above | DFCI, UKALL |
| No induction failure events; treatment failure is an event if CR has not been achieved at later timepoints | Different combinations of methods | AIEOP-BFM, ALLTogether, ALL-IC-BFM, CoALL, COG, DCOG, EsPhALL-COG, NOPHO |
| Event | Methods | Study group |
|---|---|---|
| Induction failure (EOI) ≥5% blasts in BM | Cytomorphology and/or FCM-/PCR-MRD or genetics | JCCG, SEHOP-PETHEMA, SFCE, SJCRH, TPOG |
| Induction failure (EOI) ≥25% blasts in BM | See above | DFCI, UKALL |
| No induction failure events; treatment failure is an event if CR has not been achieved at later timepoints | Different combinations of methods | AIEOP-BFM, ALLTogether, ALL-IC-BFM, CoALL, COG, DCOG, EsPhALL-COG, NOPHO |
BM: bone marrow, EOI: end of induction, FCM-MRD: flow cytometric minimal residual disease, MRD: minimal residual disease, PCR-MRD: polymerase chain reaction minimal residual disease.
Defining treatment time points
A uniform definition of the time points end of induction (EOI; approximately 4 weeks from start of treatment) and end of consolidation (EOC; approximately 8-12 weeks from start of treatment) appears feasible. Later time points, such as end of intensification or end of reintensification are also potentially useful, because the results of assessment at these time points may be used as criteria for intensification of therapy or pursuit of alternative rescue therapies.
Defining TF based on treatment time points
Some study groups consider failure to achieve CR at EOI as a TF event for EFS analysis, whereas others do not: instead, they use the result to stratify the patient to more intensive postinduction therapy, and a TF event only occurs if CR is not achieved at a later time point. This is an important limitation when trying to compare the results of different trials, because identical end-of-induction evaluations could lead to different EFS results for different protocols. For example, JCCG, SFCE, SEHOP-PETHEMA, SJCRH, and TPOG consider M2 or more blasts in BM at EOI as a TF event. In COG trials, on the other hand, a TF event occurs only if CR is not achieved after consolidation therapy (EOC). For UKALL and DFCI, M3 marrow at EOI is considered a TF event. In AIEOP-BFM ALL, ALL-IC BFM, CoALL, DCOG, and ALLTogether, lack of CR at EOI has an impact on subsequent risk stratification and intensification of postinduction treatment; however, this does not define a TF event. In these groups, a TF event is defined as lack of CR at various later time points.
Consensus for CR and TF
▪ Each study group should define the decisive treatment points EOI and EOC (and, if useful for the individual protocol, end of intensification phase) for their protocols.
▪ Several methods are available to assess MRD in ALL, including quantitative polymerase chain reaction of rearranged immunoglobulin/T-cell receptor genes using either standard allele-specific oligonucleotide PCR (ASO-PCR) or next-generation sequencing (NGS) technologies, reverse transcriptase-PCR of fusion transcripts, and FCM. All these methods are acceptable as long as standardized evaluation criteria (quality standards) have been established.
▪ Definition of CR:
• CR is to be assessed no earlier than EOI
• For CR, the following is required:
▪ BM: MRD <1% and/or M1 cytomorphology
▪ CNS: CNS1
▪ Testes: normalization of clinical examination, or a negative biopsy if clinical examination is not considered normal.
▪ EM: no evidence of leukemic infiltrates as evaluated clinically and by imaging; a preexisting leukemic mass (mediastinal mass included) must have decreased at least to one-third of the initial tumor volume
▪ CR assessment of BM should be performed by a standardized MRD method and cytomorphology. The consensus was that MRD measurement is now the ‘gold standard’ to assess CR in addition to its established role in risk stratification at the end of induction. If MRD is not available, the response is assessed by cytomorphology. If both methods are available, MRD generally takes precedence over cytomorphology. Complementary methods including genetic analysis may be used to verify CR achievement. CNS status should be based on CSF cytomorphology (other methods such as FCM or genetic analysis may be used in unclear cases), and clinical neurological examination, and CNS imaging in case of neurological clinical findings. Physical examination, imaging or histologic examination of a tissue biopsy should be used for the evaluation of non-CNS EM disease.
▪ We propose that every study group report CR status on early time points such as end of induction or end of consolidation to improve comparability between trials.
▪ Definition of TF:
• Failure to achieve CR at a clearly predefined time point (EOI, EOC, or other time points during intensification) should be considered as a TF event, and this time point should be specified at the onset of the clinical trial.
• There was progress toward a consensus that a TF event is to be defined no earlier than the EOC, because this would allow a patient not achieving CR by EOI to be offered a consolidation therapy with agents not given during induction in an effort to overcome blast resistance and potentially achieve CR. If such a patient still does not achieve CR after this risk-adapted consolidation phase, this could reasonably define a TF event.
Relapse
An overview and classification of current relapse definitions was prepared (supplemental Table 2). Obviously, a patient can only have a relapse event after a CR has previously been achieved. Relapse should be subclassified by anatomic sites involved at the time of relapse: isolated BM relapse, isolated CNS relapse, isolated testicular relapse, isolated other EM relapse involving other anatomical sites, and various combinations of these sites.
Historical definitions of relapse
Traditionally, isolated BM relapse has been defined as a M3 marrow occurring after CR with no other sites involved, with cytomorphology as the gold standard for defining a BM relapse. Combined relapse has been defined as reappearance of lymphoblasts in the BM with a threshold of ≥5%, with the involvement of one or more EM sites confirmed to be leukemic by cytology/histology or imaging. Isolated CNS relapse is defined as CNS3 status (with a M1 marrow) emerging after CR. A testicular relapse is defined as uni- or bilateral testicular enlargement with leukemic infiltration confirmed by biopsy (required by most groups) or imaging. Defining relapse in other EM anatomic sites (one and more) requires biopsy (supplemental Table 3).
Change in the threshold of leukemic blasts in BM with a suspected isolated BM relapse
Highly sensitive methods to detect leukemic cells have prompted all study groups except JCCG and SEHOP-PETHEMA to lower the threshold of morphologically defined blast percentage required for BM relapse if additional conditions are fulfilled. Thus, AIEOP-BFM, ALL-Together, CoALL, COG, DCOG, DFCI, EsPhALL/COG, NOPHO, SFCE, SJCRH, and UKALL define BM relapse as ≥5% lymphoblasts if confirmed to be leukemic by 1 (ALLTogether, CoALL, COG, DCOG, DFCI, EsPhALL/COG, NOPHO, SJCRH, UKALL) or 2 (AIEOP-BFM, SFCE) other detection methods such as PCR-MRD, FCM-MRD, NGS-MRD, or genetics. ALLTogether and COG define BM relapse as <5% (but >1%) blasts by morphology in BM if 2 additional methods indicate the re-emergence of leukemia. If only 1 sensitive diagnostic test is available, a repeat BM evaluation is required, with additional conditions to be met for this second BM assessment. It is important to emphasize that all groups agreed that relapse in the absence of a M3 morphologic result must be confirmed by specified validated diagnostic methods.
Special features of CNS relapse
All study groups accept a single lumbar puncture (LP) with CNS3 status as a relapse event. COG, DCOG, DFCI, SJCRH, and UKALL also incorporate a definition of CNS relapse that does not require CNS3. Patients with a single LP with CNS2 status after attaining CR require a second LP to be performed 1 to 4 weeks later. If this LP again shows CNS2, COG, DCOG, SJCRH, and UKALL define this to be a CNS relapse if the presence of leukemic blasts is confirmed by FCM-MRD, FISH, and/or indirect nuclear terminal deoxynucleotidyl transferase immunofluorescence assay. DFCI does not require a confirmatory test. ALLTogether and CoALL require a confirmatory test also when CNS3 is detected.
Consensus for relapse
Consensus criteria for relapse definition in pediatric ALL: BM relapse (MRD available)
| BM 1 | BM 2* | |
|---|---|---|
| MRD | Others | MRD |
| ≥25% | Not necessary to define relapse | Not necessary to define relapse |
| Five to <25% | One other test‡ with ≥1% blasts | Not necessary to define relapse |
| Five to <25% | None | Two tests† with with ≥1% blasts |
| One to <5% | Two other tests† with with ≥1% blasts | Not necessary to define relapse |
| One to <5% | Zero or 1 other test‡ with ≥1% blasts | Two tests† with ≥1% blasts |
| BM 1 | BM 2* | |
|---|---|---|
| MRD | Others | MRD |
| ≥25% | Not necessary to define relapse | Not necessary to define relapse |
| Five to <25% | One other test‡ with ≥1% blasts | Not necessary to define relapse |
| Five to <25% | None | Two tests† with with ≥1% blasts |
| One to <5% | Two other tests† with with ≥1% blasts | Not necessary to define relapse |
| One to <5% | Zero or 1 other test‡ with ≥1% blasts | Two tests† with ≥1% blasts |
Second bone marrow evaluation ≥1 wk later.
FCM/PCR/NGS-based MRD or FISH/karyotype/PCR demonstrating leukemia-specific marker or M2/M3 morphology.
Consensus criteria for relapse definition in pediatric ALL: BM relapse (MRD unavailable)
| BM 1 | BM 2* | |
|---|---|---|
| Cytomorphology | Others | Cytomorphology |
| M3 | Not necessary to define relapse | Not necessary to define relapse |
| M2 | One other test‡ with ≥1% blasts | Not necessary to define relapse |
| M2 | None | M2 |
| M1 | Two other tests† with ≥1% blasts | Not necessary to define relapse |
| BM 1 | BM 2* | |
|---|---|---|
| Cytomorphology | Others | Cytomorphology |
| M3 | Not necessary to define relapse | Not necessary to define relapse |
| M2 | One other test‡ with ≥1% blasts | Not necessary to define relapse |
| M2 | None | M2 |
| M1 | Two other tests† with ≥1% blasts | Not necessary to define relapse |
Second bone marrow evaluation ≥1 wk later.
FISH/karyotype/PCR demonstrating leukemia-specific marker.
Consensus criteria for relapse definition in pediatric ALL: CNS relapse
| CSF 1 | CSF 2* | |
|---|---|---|
| Cytomorphology | Cytomorphology | Others |
| CNS3† | Not necessary to define relapse | Not necessary to define relapse |
| CNS2 | CNS2 | One other positive test |
| CSF 1 | CSF 2* | |
|---|---|---|
| Cytomorphology | Cytomorphology | Others |
| CNS3† | Not necessary to define relapse | Not necessary to define relapse |
| CNS2 | CNS2 | One other positive test |
Second CSF evaluation ≥1 wk later.
May be defined by cytomorphology, imaging, or biopsy.
▪ The diagnosis of relapse can only be made if CR has been previously achieved.
▪ BM:
• For the purposes of defining bone marrow relapse, the percentage of blasts in any marrow evaluation should be determined using a validated MRD technique if available, with cytomorphology used only as a backup.
• To define bone marrow relapse, there must be clear evidence that the patient has ≥1% leukemic blasts in the bone marrow. The specific conditions that must be met to provide clear evidence of ≥1% leukemic blasts in the bone marrow include the following:
▪ If MRD testing is available, then a single marrow is sufficient to define relapse if MRD is ≥25%; or MRD is ≥5% with 1 additional sensitive diagnostic test demonstrating ≥1% blasts (FCM-/PCR-/NGS-MRD or FISH or cytogenetics or RT-PCR of leukemic specific marker or M2/M3 morphology); or MRD is ≥1% with 2 additional sensitive diagnostic tests demonstrating ≥1% blasts.
• If the MRD is ≥1% but without the required confirmatory tests, then a relapse can still be defined if a consecutive marrow evaluation separated by ≥1 week demonstrates ≥1% using 2 sensitive diagnostic tests.
▪ If MRD testing is unavailable, then a single marrow is sufficient to define relapse if M3 morphology or M2 morphology with 1 additional sensitive diagnostic test demonstrating ≥1% blasts (FISH or cytogenetics or RT-PCR of leukemic specific marker) or M1 morphology with 2 additional sensitive diagnostic tests demonstrating ≥1% blasts is present.
• If the morphology is M2 with no confirmatory tests, then a relapse can still be defined if a consecutive marrow evaluation separated by ≥1 week demonstrates M2 morphology.
▪ CNS:
• CNS3 (by cytomorphology or imaging or biopsy)
• CNS2 on 2 consecutive cytomorphologic evaluations at least 1 week apart with 1 additional consistent and sensitive diagnostic test present in ≥1 of the evaluations
▪ Other EM sites:
• EM tumor burden, confirmed by imaging and/or biopsy. Imaging could be diagnostic if combined with other sites, but a positive biopsy is needed to define relapse if it is the only potential relapse site
▪ Combined relapse:
• any BM relapse combined with any non-BM relapse (with each defined above)
The immediate impact of this newly agreed on consensus is illustrated by an example in Figure 1.
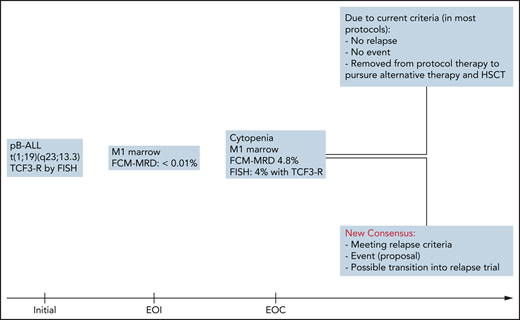
Differences and consequences of current vs new consensus relapse criteria: a case study. HSCT, hematopoietic stem cell transplantation; pB-ALL, precursor B-acute lymphoblastic leukemia; TCF3-R, transcription factor 3 rearrangement.
Differences and consequences of current vs new consensus relapse criteria: a case study. HSCT, hematopoietic stem cell transplantation; pB-ALL, precursor B-acute lymphoblastic leukemia; TCF3-R, transcription factor 3 rearrangement.
Discussion
Establishing common criteria for response and relapse assessment in pediatric ALL is urgently needed, and the PDL group believes that these criteria should be adopted by all study groups. The implementation of new consensus criteria will entail significant additional effort for most study groups. Amendments to the respective protocols might be required, or it may be necessary to wait until the next clinical trial is implemented. Obviously, any prioritization techniques to determine slow response, treatment failure, and/or relapse will depend on the availability of such diagnostic tests.
Currently, crucial end points in clinical trials are poorly defined. This inhibits consistency not only in the definition of EFS but also in eligibility criteria to enroll patients into trials for relapsed or refractory ALL. This creates major challenges in comparing the efficacy of the treatments in such trials. Uniform eligibility for trials for relapsed or refractory ALL may facilitate seamless transition from upfront to second-line trials. It may also reduce the proportion of patients who are taken off protocol as they previously did not qualify for a subsequent clinical trial.
It appears no longer appropriate to require identification of ≥25% BM blasts by morphology to define relapse, when an impending relapse can be detected using sensitive, objective, highly reproducible, and well-standardized MRD methods.7,39,40 The results of trials that do not adopt these response definitions will be confounded if patients are being removed from protocol therapy without meeting a protocol-specified event, which complicates EFS comparisons. For example, individual clinicians, patients, and their families may be unwilling to continue frontline therapy in the face of clear evidence of recurrent disease that is not defined as relapse in the treatment protocol. Such patients may reasonably pursue alternative therapies (eg, chimeric antigen receptor T cells or other immunotherapeutic approaches) before morphologic relapse, especially because recent data suggest that such therapies are more likely to be safe and effective in the MRD setting than following an overt morphologic relapse.41,42 Indeed, patients may benefit from more timely initiation of alternative salvage therapy. Particularly in the context of clinical trials in relapsed or refractory ALL, uniform eligibility criteria are crucial to compare the efficacy of alternative salvage therapies. Clear definitions for CR, TF, relapse, and EFS will be also useful for regulatory agencies in evaluating applications for approval of new drugs or therapies or by expansion of the indication for an approved drug or therapy.
During the work for this project, it was repeatedly emphasized that an actionable threshold for each diagnostic method should be carefully considered, and although the threshold may differ among various methods, the results of FCM- and PCR-MRD are very consistent.43 This aspect is important because the sensitivity for detection and quantification depends on the method itself, and availability of each method may vary between study groups. For example, for most probes, the sensitivity of FISH testing ranges from 1% to 5%, whereas for FCM-MRD, PCR-MRD, and NGS-MRD, much lower thresholds of disease detection are possible. Even within a given methodology, however, it is extremely difficult to set uniform threshold levels for diagnostic tests in all study groups because individual assays may have different validated sensitivity thresholds. We provide a supplemental Appendix comprising suggestions how thresholds may be defined for different scenarios of test availability.
The authors acknowledge that the MRD technologies used to define response and relapse may not be widely available in low- and middle-income countries. In this case, standard morphologic definitions should be used. However, there is also an urgent need to establish standardized and affordable MRD technologies to more sensitively detect patient responses and improve treatment outcomes.
In summary, the efforts of the PDL group to establish uniform definitions of CR, TF, and relapse among the major international ALL consortia, provided insights into the adaptations that individual groups have made with the ever-advancing technologies to detect microscopic disease in the era of molecular diagnostics. Although it was not possible for the groups to agree on every aspect, important progress was made to come to consensus on major aspects of these definitions. This will facilitate a more uniform interpretation of treatment outcomes in newly diagnosed patients, and perhaps even more importantly, about the benefit of novel therapeutic interventions in patients for whom upfront therapies fail.
Acknowledgments
The authors thank Julia Alten, Isabel Badell, Claus R. Bartram, Giuseppe Basso, Monika Brüggemann, Mireia Camós, Hélène Cavé, Valentino Conter, José Luis Dapena, Michael Dworzak, Eva Fronkova, Hester de Groot-Kruseman, Renate Panzer-Grümayer, Oskar Haas, Valerie de Haas, Jochen Harbott, Ondrej Hrusak, Janos Kappelmayer, Der-Cherg Liang, Wolf-Dieter Ludwig, Eva Mejstrikova, Anja Möricke, Manuel Ramírez, Jose M. Ribera, Brigitte Schlegelberger, Martin Stanulla, Maria Grazia Valsecchi, Jan Trka, Marketa Zaliova, Martin Zimmermann, and Jan Zuna for their valuable contribution and input.
This project was supported by Deutsche Krebshilfe grants 108588, 70112517, and 70112958, the Italian Association for Cancer Research (AIRC IG 2017: 20564, AIRC IG 2018: 21724 and IG2015:17593, CRUK/AIRC/FC AECC 22791, AIRC Molecular Clinical Oncology 5 per mille 21147), the French Hospital Program for Clinical Research (PHRC 10-02-05), the Danish Childhood Cancer Foundation (2019-5934), the Danish Cancer Society (R257-A14720), US National Cancer Institute (CA021765), American Lebanese Syrian Associated Charities, the Swedish Childhood Cancer Fund (TJ2017-0076), Japan Agency for Medical Research and Development (AMED) (21ck0106612h0002), Blood Cancer UK, NCTN Operations Center and Statistics & Data Center (SDC) grants (U10CA180886 and U10CA180899, respectively) and the St Baldrick's Foundation (COG).
Authorship
Contribution: M.S. and P.B. initiated the topic; S.B. and M.S. wrote the paper; and A. Baruchel, A. Biondi, M.B., M.C., G. Cario, G. Cazzaniga, G.E., C.J.H., M.H., S.P.H., C.K., H.-C.L., F.L., M.L.L., A.M., G.M., R.P., C.-H.P., S.R., K.S., L.B.S., J.S., A.V., and P.B. discussed the consensus and reviewed the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Martin Schrappe, Department of Pediatrics, University Medical Center Schleswig-Holstein, Campus Kiel, Arnold-Heller-Str. 3, Bldg C, 24105 Kiel, Germany; email: m.schrappe@pediatrics.uni-kiel.de.
The online version of this article contains a data supplement.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal