

In this issue of Blood, Chen et al identified the intestinal IL-33–ST2–serotonin axis as a key immune-endocrinal crosstalk that drives platelet reactivity during hemostasis and platelet-dependent neutrophil recruitment during inflammation.1
The gut is the largest endocrine organ in the body able to sense environmental cues to regulate intestinal homeostasis and host defense. A breakdown in the intestinal homeostasis can lead to inflammatory bowel diseases (IBDs), such as Crohn’s disease and ulcerative colitis, which are associated with increased platelet reactivity and increased susceptibility to thromboembolic complications.2
The intestinal epithelium is a heterogeneous tissue comprised of multiple cell types, including neuroendocrine and intestinal epithelial cells (IECs). Enterochromaffin (EC) cells are a subset of intestinal enteroendocrine cells with potent mechano-sensors that are able to translate environmental signals to produce endocrine molecules such as the neurotransmitter and hormone serotonin (5-hydroxytryptamine [5-HT]). More recently, immune-driven signals released from IECs were also shown to stimulate the secretion of 5-HT from EC cells.3 In response to intestinal damage or stress, IECs secrete cytokines such as interleukin-33 (IL-33), regulating intestinal homeostasis and immune responses.
IL-33, a member of the IL-1 family, is constitutively expressed in endothelial, stromal, and epithelial cells. IL-33 is present in the gut in steady state and its expression and release from IECs are increased during cell necrosis and tissue damage. In contrast to cell necrosis, IL-33 is inactivated during cell apoptosis by caspases, which limits an inflammatory response during programmed cell death. IL-33 acts as an alarmin by binding to its receptor ST2 expressed on hematopoietic and nonhematopoietic cells and participates in mucosal homeostasis and host defense. IL-33 supports allergic responses on multiple barrier sites, including the intestinal epithelium, by promoting type 2 immune response.4
Recently, Chen et al identified IL-33 released from necrotic IECs as a key trigger for quick serotonin release from EC cells, leading to gut mobility and peristaltic movement which facilitates the expulsion of parasites.3 EC cells are the major source of peripheral 5-HT, and the release of 5-HT supports epithelial secretion, platelet function, peristalsis, wound healing, neutrophil recruitment to the site of inflammation,5 and mucosal immunity. Serotonin synthesis is catalyzed by the enzyme tryptophan hydroxylase (TPH) with more than 90% of the released 5-HT taken up by platelets through the 5-HT transporter (SERT), then stored in dense granules and released upon platelet activation. Increased serotonin levels are associated with cardiovascular diseases and inflammatory diseases associated with elevated risk of thrombosis,6 making this axis an interesting target for limiting thrombo-inflammation. Understanding the contribution of the gut IL-33–5-HT axis to hemostasis and inflammation provides important insights to its relevance and its potential therapeutic benefit in a disease states associated with thrombosis.
In their article, Chen et al addressed the effect of IL-33 on platelet reactivity, hemostasis, wound healing, and inflammation in mice. By using IL-33–deficient mice, the authors showed an increase in tail bleeding time compared to control mice, which was associated with compromised platelet response to stimulation. This effect was due to the absence of IL-33 secretion from IECs, limiting 5-HT release from EC cells. The defect in 5-HT secretion results in a reduction of serum 5-HT and altered platelet response. A recent study by Chen et al3 showed that IL-33–ST2 signaling induces PLC-γ1 activation and increased intracellular Ca2+-dependent 5-HT secretion from EC cells, independent of the classical MyD88 signaling downstream of ST2. Exploration of a therapeutic strategy showed that perfusion of IL-33 did not induce platelet activation in vivo, but it did increase platelet reactivity to thrombin ex vivo. These results are in line with previous data showing that 5-HT is a helper agonist that synergizes with other agonists such as adenosine 5′-diphosphate (ADP) to potentiate platelet activation.7 Therefore, in the absence of damage, constitutive IL-33 is important for 5-HT release supporting platelet reactivity during hemostasis.
Next, the authors assessed the relevance of gut IL-33 in the context of intestinal damage, as observed in dextran sulfate sodium (DSS)–induced colitis and thioglycollate-induced sterile peritonitis (see figure). In both models, IL-33 levels in the colon were increased after induction of inflammation associated with increased serum 5-HT. Inhibition of intestinal IL-33 reduced 5-HT serum levels and decreased platelet reactivity. The authors investigated the relevance of increased platelet reactivity on neutrophil recruitment in a skin wound healing model and 2 inflammatory models: thioglycollate-induced sterile peritonitis and lipopolysaccharide (LPS)-mediated endotoxic shock. Platelet reactivity and neutrophil recruitment were increased in the skin wound healing and thioglycollate-induced peritonitis models. In the skin wound healing model, gut IL-33–mediated platelet reactivity and neutrophil recruitment accelerated wound healing due to increased neutrophil infiltration in early wound healing facilitating the elimination of debris. It is also possible that increased hemostasis resulting from platelet hyperreactivity can contribute to accelerated wound healing. Deletion of IL-33 from IECs or Tph-1 from EC cells inhibits serotonin release, platelet reactivity, and neutrophil recruitment during sterile peritonitis. In a model of LPS-induced endotoxic shock, IL-33–mediated release of 5-HT supported acute neutrophil recruitment, inflammatory cytokine release, and increased mortality. Deletion of IL-33 in IECs (or Tph-1 in EC cells) reduced neutrophil recruitment, and this defect was restored by injection of recombinant IL-33 or 5-hydroxytryptophan (5-HTP). This effect was dependent on platelet serotonin as injection of fluoxetine (a selective serotonin reuptake inhibitor) or platelet depletion inhibited neutrophil recruitment. Indeed, the absence of platelet serotonin improved survival in LPS-induced endotoxic shock model. Moreover, neutrophil extravasation into the lung, peritoneum, and skin wounds was reduced in Tph1-deficient mice.5 Together, these results show that gut IL-33 supports platelet reactivity and neutrophil recruitment via serotonin release. However, targeting this axis to improve disease outcome will require further investigation. In a mouse model of cecal ligation and puncture-induced sepsis, IL-33 was shown to promote recruitment of neutrophils, which resulted in increased bacterial clearance and decreased mortality. In parallel, this study shows that IL-33–induced hyperreactive platelets potentiate neutrophil recruitment and mortality in LPS-mediated endotoxic shock.
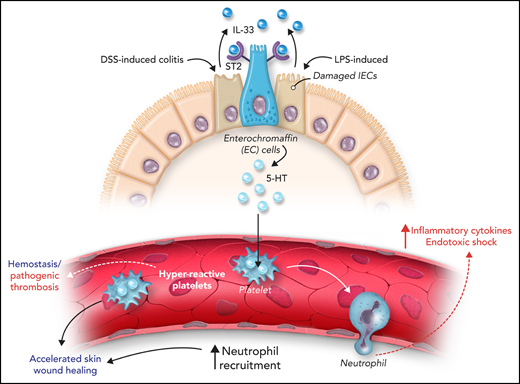
Intestinal IL-33 increases platelet reactivity, hemostasis, and inflammation via serotonin release. Constitutive IL-33 secretion from IECs supports serotonin (5-HT) release from enterochromaffin cells. 5-HT is taken up by platelets, increasing platelet reactivity, hemostasis, and wound repair. After intestinal inflammation induced by DSS or LPS, IL-33 release from damaged IECs supports 5-HT secretion and platelet-dependent neutrophil recruitment.
Intestinal IL-33 increases platelet reactivity, hemostasis, and inflammation via serotonin release. Constitutive IL-33 secretion from IECs supports serotonin (5-HT) release from enterochromaffin cells. 5-HT is taken up by platelets, increasing platelet reactivity, hemostasis, and wound repair. After intestinal inflammation induced by DSS or LPS, IL-33 release from damaged IECs supports 5-HT secretion and platelet-dependent neutrophil recruitment.
In conclusion, this work by Chen et al described a new gut endocrine-immune axis that regulates platelet reactivity in hemostasis and inflammation. As IL-33 levels are increased in inflamed colonic biopsies from patients with IBDs,8 IL-33 may represent a potential therapeutic route for reducing both platelet reactivity and neutrophil recruitment, which would limit thrombo-inflammation. The Chen et al study also opens new possibilities in other conditions associated with thromboembolic conditions like those observed in trauma patients and patients undergoing chemotherapy. However, the beneficial vs detrimental effects of targeting this axis have not yet been determined. In which pathogenic condition and at what stage of disease development and progression would the IL-33–ST2–5-HT axis be a beneficial target? Could IL-33 become a therapy in bleeding disorders? Future work exploring these possibilities might recognize IL-33 as a novel regulator of hemostasis and thrombo-inflammation.
Conflict-of-interest disclosure: The author declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal