

Key Points
Intestinal IL-33 stimulates platelet function by enhancing 5-HT release, promoting neutrophil recruitment during acute inflammation.

Abstract
Peripheral serotonin (5-HT) is mainly generated from the gastrointestinal tract and taken up and stored by platelets in the circulation. Although the gut is recognized as a major immune organ, how intestinal local immune responses control whole-body physiology via 5-HT remains unclear. Here, we show that intestinal inflammation enhances systemic platelet activation and blood coagulation. Intestinal epithelium damage induces elevated levels of the alarm cytokine interleukin-33 (IL-33), leading to platelet activation via promotion of gut-derived 5-HT release. More importantly, we found that loss of intestinal epithelial-derived IL-33 lowers peripheral 5-HT levels, resulting in compromised platelet activation and hemostasis. Functionally, intestinal IL-33 contributes to the recruitment of neutrophils to sites of acute inflammation by enhancing platelet activities. Genetic deletion of intestinal IL-33 or neutralization of peripheral IL-33 protects animals from lipopolysaccharide endotoxic shock through attenuated neutrophil extravasation. Therefore, our data establish a distinct role of intestinal IL-33 in activating platelets by promoting 5-HT release for systemic physiology and inflammation.
Introduction
Enterochromaffin (EC) cells as a subtype of enteroendocrine cells on the intestinal epithelium are known as the main source of peripheral serotonin (5-HT).1 Mucosal 5-HT is considered to function as both a proinflammatory and an anti-inflammatory molecule during intestinal inflammation.2-4 In addition to its presence in the gut, the majority of EC cell–derived 5-HT can also be taken up and stored in circulating platelets at rest and released upon activation.5,6 5-HT is critical for the platelet function of hemostasis, considering its constant surveillance of the endothelium for vascular integrity.7,8 In addition to its role during hemostatic response and wound repair, platelet 5-HT is also critical for the recruitment of leukocytes, mainly neutrophils, to the affected site after tissue injury or infection.9-12 Therefore, peripheral 5-HT exerts its function via participating in both intra- and extraintestinal reflexes to regulate body physiology.
It is now known that the gastrointestinal (GI) tract does not function independently, but instead requires dynamic interaction with other organ systems. Accordingly, the state of the gut largely influences the function of other compartments in the body.13 Clinical studies indicate that in more than one-third of patients, inflammatory bowel disease (IBD) is associated with different extraintestinal manifestations beyond the original GI symptom, related to nearly every organ.14 Thromboembolism is an IBD-dependent extraintestinal manifestation with high morbidity and mortality.15 Patients with IBD in both active and remission phases display higher levels of blood clotting and fibrinolysis than the general population.16 Although inflammation and damage in the colonic tissues have been implicated in contributing to thromboembolism, the precise mechanisms for such extraintestinal manifestation are still unclear.
Interleukin-33 (IL-33), an IL-1 family member, is constitutively expressed and released from barrier surfaces, such as intestinal epithelial cells, and functions as an alarm cytokine.17 Although IL-33 is likely to be required for tissue homeostasis because of its constitutive expression in a physiological state,18 excessive IL-33 release after tissue damage following trauma or infection could also lead to various of pathologic consequences.19-23 IL-33 has been reported to play an important role in intestinal development and homeostasis.24,25 However, whether and how intestinal IL-33 affects extraintestinal physiology and pathophysiology remain unclear. In the current study, we discovered that intestinal injury elevates colonic IL-33 levels, which in turn stimulates 5-HT release from EC cells. Consequently, it promotes platelet activation and blood clotting. Furthermore, loss of intestinal IL-33 signaling represses neutrophil recruitment during acute inflammation through dampened platelet 5-HT. Our data illustrate that intestinal immune-endocrinal crosstalk plays a key role in regulating systemic physiology and inflammation by modulating platelet function.
Materials and methods
Animals
C57BL/6 (wild-type [WT]) and Vil1Cre mice were purchased from The Jackson Laboratory; Il33−/− mice were from Susumu Nakae; Tph1flox mice were from Gerard Karsenty; Il1rl1flox and Il33flox mice were from the Knockout Mouse Project; ChgaCreER mice were from the European Mouse Mutant Archive; Tph1CFP mice were from Andrew B. Leiter; Il1rl1−/− mice were from Giorgio Trinchieri. Mice were maintained at the National Cancer Institute facilities under specific pathogen-free conditions. Mouse lines were interbred in our facilities to obtain the final strains described in the text. Mice were fed a standard chow diet, and most experiments were conducted at age 7 to 12 weeks. All experiments were carried out in accordance with guidelines prescribed by the Institutional Animal Care and Use Committee at the National Cancer Institute. For ChgaCreERIl1rl1fl/fl mice, 1 mg of tamoxifen (Sigma-Aldrich) was intraperitoneally (IP) injected daily for 5 days before the experiments.
DSS-induced colitis model
Experimental colitis was initiated by treatment of mice with 2.5% dextran sulfate sodium (DSS; 36 000-50 000 molecular weight; MP Biomedicals) in drinking water for 6 days. DSS was then replaced with normal water for another ∼6 to 7 days. Body weight was monitored daily.
Tail bleeding assay
Mice were anesthetized with a mixture of ketamine, xylazine, and acepromazine and placed in a prone position. A distal 10-mm segment of the tail was amputated with a scalpel. The tail was immediately immersed in a 50-mL Falcon tube containing isotonic saline prewarmed in a water bath to 37°C. Each animal was monitored for 20 minutes, even if bleeding ceased, to detect any rebleeding. Bleeding time was determined using a stop clock. If bleeding on/off cycles occurred, the sum of bleeding times within the 20-minute period was used. The experiment was terminated at the end of 20 minutes to avoid lethality during the experiment.
Blood cell count
Blood was collected by cardiac puncture into EDTA-containing tubes. The numbers of complete blood count, including red blood cells, white blood cells, and platelets, were read on a HemaVet 950 analyzer by the Molecular Histopathology Laboratory at the National Cancer Institute.
Platelet analysis
Platelets were purified from mouse plasma and resuspended with Tyrode’s buffer (134 mM of sodium chloride, 0.34 mM of disodium phosphate, 2.9 mM of potassium chloride, 12 mM of sodium bicarbonate, 20 mM of N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid [pH 7.0], 5 mM of glucose, and 0.35% bovine serum albumin). For flow cytometric analysis, antibodies were added: anti-CD42b, anti-CD62P and Jon/A (antiactivated CD41/CD61; Emfret), or anti-ST2 (BioLegend). For platelet activation, thrombin (Sigma-Aldrich) was added at 0.05 U/mL or at the indicated dose together with the antibodies and incubated at room temperature for 15 minutes. Phosphate-buffered saline (PBS) was added to stop the reaction, and platelets were analyzed by fluorescence-activated cell sorting within 30 minutes. All flow cytometric data were acquired on a BD X-20 cell analyzer and analyzed with FlowJo software.
Serotonin measurements
For serotonin analysis, blood samples were collected by cardiac puncture, and serum or platelets were isolated. Measurements were performed using the Serotonin ELISA Kit (Abnova) according to the manufacturer’s protocol.
Sterile peritonitis
Adult mice were treated with 1 mL of 4% thioglycollate (Sigma-Aldrich) IP for chemical irritation. For IL-33 treatment, 200 ng of IL-33 (BioLegend) was injected IP 10 minutes before the treatment. After 4 hours, the mice were euthanized, and cells were recovered by peritoneal lavage. Peritoneal cells were washed with PBS and stained with antibodies anti-Ly6G and anti-F4/80 (BioLegend) for flow cytometric analysis. For platelet depletion, the mice were injected with 2 μg/g of anti-CD42b antibody or rat immunoglobulin G control (Emfret) IV for 24 hours. The mice were then treated for peritonitis 10 minutes after PBS or 200 ng of IL-33 treatment. For fluoxetine treatment, the mice were injected with 40 mg/kg of fluoxetine (Sigma-Aldrich) IP for 2 hours. The mice were then treated for peritonitis 10 minutes after PBS or 200 ng of IL-33 treatment. The Il33−/− mice were given 1.5 mg/mL of 5-HTP (Sigma-Aldrich) in drinking water for 2 weeks, and the mice were treated with fluoxetine injection. After 2 hours, the mice were then induced for peritonitis.
Aseptic wound healing
The mice were anesthetized with a mixture of ketamine, xylazine, and acepromazine, and hair on the back was removed with an electric razor. A sterile 4-mm biopsy punch was used to outline 2 circular patterns for the wound on either side of the mouse midline at the level of the shoulders. The wounds were made by picking up a fold of skin and punching through the 2 layers of skin. Wounds were monitored and photographed daily until 2 weeks, and buprenorphine SR was injected subcutaneously every 2 days until a scab developed at the wound site. The diameter of each wound was then measured using ImagJ to calculate the percentage of wound closure compared with the day-0 wound diameter. For histology, the skin including wound was cut off 4 hours after injury induction. The skin was fixed in 10% neutral formalin and stored in 70% ethanol. Samples were then paraffin embedded and cut into 10-μm sections, and hematoxylin and eosin staining was performed by Histoserv (Germantown, MD). For myeloperoxidase (MPO) activity determination, samples were washed and homogenized, and tested was performed using the MPO Fluorometric Activity Assay Kit (Sigma-Aldrich) according to the manufacturer’s protocol. For IL-33 treatment, 200 ng of IL-33 (BioLegend) was IP injected 10 minutes before the surgery.
LPS endotoxic shock
Adult male mice were challenged with an IP injection of Escherichia coli serotype 055:B5 lipopolysaccharide (LPS; Sigma-Aldrich) at a dose of 20 mg/kg. The mice were euthanized when they exhibited q of the following clinical signs: >4°C temperature drop, severely labored breathing, somnolence, paralysis, bleeding, sustained severe tremor, lack of righting reflex, or morbidity. For flow cytometric analysis, the mice were euthanized 4 hours after LPS injection, and cells were recovered by peritoneal lavage. Peritoneal cells were washed with PBS and stained with antibodies anti-Ly6G and anti-F4/80 (BioLegend). For IL-33 treatment, 200 ng of IL-33 (BioLegend) was IP injected 10 minutes before LPS injection.
Soluble ST2 treatment
pcDNA3.1-sST2-Fc and pcDNA3.1-hIgG1-Fc plasmids were from Keizo Takenaga. The plasmids were midiprepped using a plasmid extraction kit (Macherey-Nagel) and eluted in 0.9% saline solution. Plasmids (10 μg) were IV injected into the mouse tail. LPS treatment was performed 24 hours after plasmid injection.
Cytokine measurement
For IL-33 quantification, colon or skin tissue after wound induction was isolated and washed with PBS. Tissue samples were weighted and homogenized in PBS. Supernatant was taken for measurement using an IL-33 ELISA kit (Invitrogen) according to the manufacturer’s instructions.
For interferon-γ, tumor necrosis factor-α, and IL-6 quantification, mouse serum was purified 24 hours after LPS treatment. Serum samples were used for cytokine measurement using the ELISA MAX Standard Set (BioLegend) according to the manufacturer’s protocol.
Statistical Analysis
Statistical analyses were performed with GraphPad Prism 5.0 (GraphPad Software, La Jolla, CA) using an unpaired 2-tailed Student t test. Statistical significance was defined as P < .05.
Results
IL-33 determines enhanced blood coagulation during intestinal inflammation
Pervious clinical studies have shown that patients with ulcerative colitis or Crohn’s disease usually also have thromboembolic disease. Anticoagulant therapy is recommended to improve the course of the intestinal inflammation.26,27 We therefore asked whether there is a causative effect of intestinal inflammation on altered blood coagulation. By using a DSS-induced colitis model (supplemental Figure 1A), we found shortened blood clotting times on days 4 and 8 after DSS treatment (Figure 1A). Platelets are known to be critical for blood coagulation.28 Although no change in platelet number was seen (Figure 1B), in vitro thrombin stimulation indicated increased platelet activation from DSS-treated animals compared with naïve mice, as measured by elevated expression of activation markers CD62P (P-selectin) and Jon/A (CD41 and CD61; supplemental Figure 1B-D; Figure 1C-D). These findings suggest that intestinal inflammation and epithelium damage promote platelet activation for blood clotting.
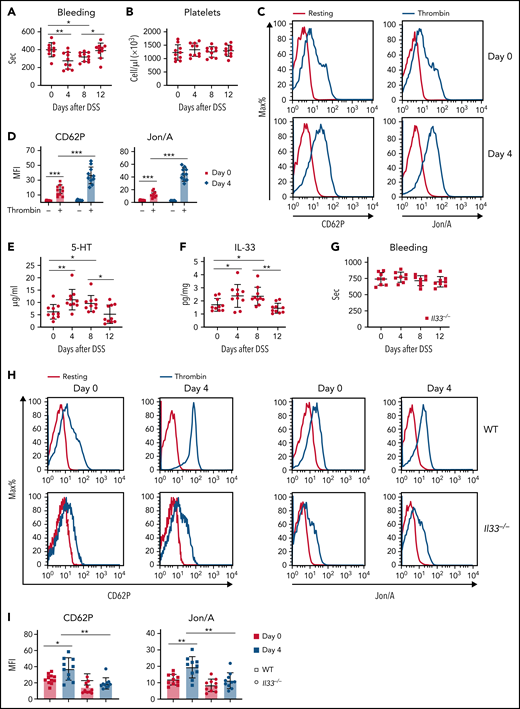
IL-33 determines enhanced blood coagulation during intestinal inflammation. (A-F) WT mice were induced with DSS colitis and examined at indicated time points. (A) Time to cessation of bleeding in response to tail injury. (B) Platelet counts during DSS colitis. (C) Representative histograms of fluorescence intensity of CD62P and Jon/A for platelets treated with or without thrombin. (D) Quantification of geometric mean fluorescence intensity (MFI) in panel C. (E) 5-HT levels in serum were assessed by ELISA. (F) IL-33 levels in colonic tissue were assessed by ELISA. (G-I) Il33−/− mice were induced with DSS colitis and examined at indicated time points. (G) Time to cessation of bleeding in response to tail injury. (H) Representative histograms of fluorescence intensity of CD62P and Jon/A for platelets treated with or without thrombin between WT and Il33−/− mice. (I) Quantification of geometric MFI in panel H. Data are representative of 2 independent experiments (C,H) or are pooled from 2 independent experiments (A-B, D-G, I). *P < .05, **P < .01, ***P < .001 by Student t test. Error bars represent standard deviation.
IL-33 determines enhanced blood coagulation during intestinal inflammation. (A-F) WT mice were induced with DSS colitis and examined at indicated time points. (A) Time to cessation of bleeding in response to tail injury. (B) Platelet counts during DSS colitis. (C) Representative histograms of fluorescence intensity of CD62P and Jon/A for platelets treated with or without thrombin. (D) Quantification of geometric mean fluorescence intensity (MFI) in panel C. (E) 5-HT levels in serum were assessed by ELISA. (F) IL-33 levels in colonic tissue were assessed by ELISA. (G-I) Il33−/− mice were induced with DSS colitis and examined at indicated time points. (G) Time to cessation of bleeding in response to tail injury. (H) Representative histograms of fluorescence intensity of CD62P and Jon/A for platelets treated with or without thrombin between WT and Il33−/− mice. (I) Quantification of geometric MFI in panel H. Data are representative of 2 independent experiments (C,H) or are pooled from 2 independent experiments (A-B, D-G, I). *P < .05, **P < .01, ***P < .001 by Student t test. Error bars represent standard deviation.
Previous studies have shown that gut-derived 5-HT is critical for platelet function.6,8 Indeed, we observed elevated levels of serum and platelet 5-HT during DSS treatment (Figure 1E; supplemental Figure 1E). Our recent work demonstrated that IL-33 promotes 5-HT release from EC cells in the gut.29 Given that DSS-mediated intestinal epithelium damage leads to IL-33 release,17 we found increased colonic IL-33 after DSS treatment (Figure 1F). We next used Il33−/− mice and noticed that loss of IL-33 abolished enhanced blood clotting during DSS colitis (Figure 1G). In addition, neither platelet counts nor serum 5-HT in Il33−/− mice were found to fluctuate after DSS treatment (supplemental Figure 1F-G). Importantly, unlike in WT mice, DSS-promoted platelet activation was largely diminished in IL-33–deficient mice (Figure 1H-I). We showed that acute intestinal injury results in enhanced blood clotting in an IL-33–dependent manner.
Intestinal IL-33–ST2 axis promotes platelet function for hemostasis
We next examined IL-33–deficient mice and found prolonged clotting time with normal platelet counts, as well as normal immune cell profiling, compared with WT mice (supplemental Figure 2A-B). We further used Vil1creIl33fl/fl mice to investigate whether intestinal epithelium-derived IL-33 is sufficient to promote systemic platelet function. Vil1creIl33fl/fl mice exhibited reduced colonic IL-33 levels compared with control animals (supplemental Figure 2C). Similar to Il33−/− mice, we found compromised platelet hemostasis in Vil1creIl33fl/fl but unchanged platelet counts (Figure 2A-B). Vil1creIl33fl/fl mice also displayed reduced serum 5-HT (Figure 2C) and platelet activation (Figure 2D-E). These data indicate that intestinal IL-33 is important for peripheral 5-HT levels and blood coagulation.
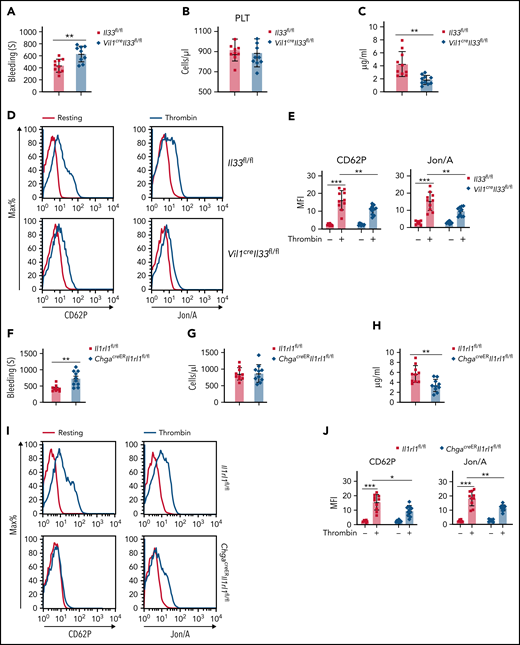
Intestinal IL-33 promotes platelet (PLT) function for hemostasis. (A) Time to cessation of bleeding in response to tail injury between Il33fl/fl and Vil1creIl33fl/fl mice. (B) PLT counts between Il33fl/fl and Vil1creIl33fl/fl mice. (C) Relative 5-HT levels in serum were assessed by ELISA between Il33fl/fl and Vil1creIl33fl/fl mice. (D) Representative histograms of fluorescence intensity of CD62P and Jon/A for PLTs treated with or without thrombin between Il33fl/fl and Vil1creIl33fl/fl mice. (E) Quantification of geometric mean fluorescence intensity (MFI) in panel D. (F) Time to cessation of bleeding in response to tail injury between Il1rl1fl/fl and ChgacreERIl1rl1fl/fl mice. (G) PLT counts between Il1rl1fl/fl and ChgacreERIl1rl1fl/fl mice. (H) Relative 5-HT levels in serum were assessed by ELISA between Il1rl1fl/fl and ChgacreERIl1rl1fl/fl mice. (I) Representative histograms of fluorescence intensity of CD62P and Jon/A for PLTs treated with or without thrombin between Il1rl1fl/fl and ChgacreERIl1rl1fl/fl mice. (J) Quantification of geometric MFI in panel I. Data are representative of 2 independent experiments (D,I) or are pooled from 2 independent experiments (A-C, E-H, J). *P < .05, **P < .01, ***P < .001 by Student t test. Error bars represent standard deviation.
Intestinal IL-33 promotes platelet (PLT) function for hemostasis. (A) Time to cessation of bleeding in response to tail injury between Il33fl/fl and Vil1creIl33fl/fl mice. (B) PLT counts between Il33fl/fl and Vil1creIl33fl/fl mice. (C) Relative 5-HT levels in serum were assessed by ELISA between Il33fl/fl and Vil1creIl33fl/fl mice. (D) Representative histograms of fluorescence intensity of CD62P and Jon/A for PLTs treated with or without thrombin between Il33fl/fl and Vil1creIl33fl/fl mice. (E) Quantification of geometric mean fluorescence intensity (MFI) in panel D. (F) Time to cessation of bleeding in response to tail injury between Il1rl1fl/fl and ChgacreERIl1rl1fl/fl mice. (G) PLT counts between Il1rl1fl/fl and ChgacreERIl1rl1fl/fl mice. (H) Relative 5-HT levels in serum were assessed by ELISA between Il1rl1fl/fl and ChgacreERIl1rl1fl/fl mice. (I) Representative histograms of fluorescence intensity of CD62P and Jon/A for PLTs treated with or without thrombin between Il1rl1fl/fl and ChgacreERIl1rl1fl/fl mice. (J) Quantification of geometric MFI in panel I. Data are representative of 2 independent experiments (D,I) or are pooled from 2 independent experiments (A-C, E-H, J). *P < .05, **P < .01, ***P < .001 by Student t test. Error bars represent standard deviation.
Loss of IL-33 receptor (Il1rl1−/−) resulted in normal blood components but prolonged blood bleeding time (supplemental Figure 2D-E). Our previous data show that IL-33 signal induces EC cell activation for rapid 5-HT release via ST2.29 Conditional deletion of ST2 from EC cells (ChgacreERIl1rl1fl/fl) led to longer bleeding time with normal platelet counts compared with controls (Figure 2F-G), along with reduced 5-HT levels in the serum (Figure 2H). Also, ChgacreERIl1rl1fl/fl mice exhibited reduced expression of CD62P and Jon/A on platelets, indicating impaired platelet activity (Figure 2I-J). These results suggest that intestinal IL-33-ST2 signaling is critical for platelet function and hemostasis.
IL-33 rapidly enhances platelet function for hemostasis
Supplementation of IL-33 led to rapid 5-HT release,29 and the uptake of 5-HT by platelets also occurred quickly.30 Shortly (10 minutes) after IL-33 injection, we noticed enhanced platelet activity in both WT and Vil1creIl33fl/fl mice (Figure 3A-D), whereas such an effect was largely abolished in ChgacreERIl1rl1fl/fl mice (Figure 3E-F), suggesting that IL-33–mediated platelet activity depends on intestinal EC cells. Accordingly, we found that IL-33 supplementation improved blood clotting in both Vil1creIl33fl/fl and control mice (Figure 3G) but failed to do so when its receptor ST2 was deleted in EC cells (Figure 3H). Additionally, ST2 was not expressed on platelets (supplemental Figure 3A), and IL-33 did not directly activate platelets (supplemental Figure 3B-C). Thus, we excluded the direct effect of IL-33 on platelets. We next used Vil1creTph1fl/fl mice to eliminate gut-derived peripheral 5-HT. Compared with WT mice (Figure 3A-B), IL-33 injection did not alter platelet activity in Vil1creTph1fl/fl mice (Figure 3I-J). Consequently, administration of IL-33 failed to shorten bleeding time in Vil1creTph1fl/fl mice (Figure 3K), suggesting that 5-HT is essential for IL-33–mediated blood clotting. Collectively, our data suggest that IL-33 induces rapid platelet activation for hemostasis by promoting EC cell–derived 5-HT release.
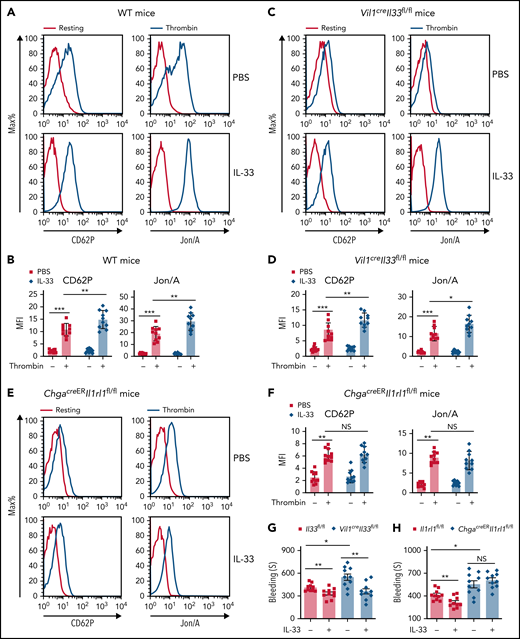
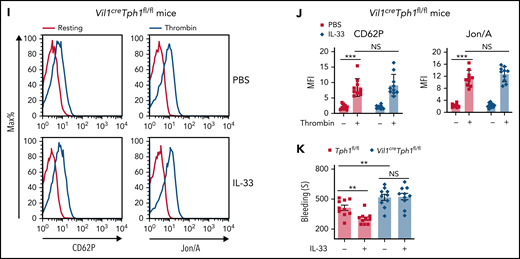
IL-33 rapidly enhances platelet function for hemostasis. (A-F) Indicated mice were injected with PBS or IL-33. Platelet activities were assessed by flow cytometry 10 minutes after injection. Representative histograms of fluorescence intensity and quantification of geometric mean fluorescence intensity of CD62P and Jon/A for platelets treated with or without thrombin from WT (A-B), Vil1creIl33fl/fl (C-D), and ChgacreERIl1rl1fl/fl (E-F) mice. (G-H) Il33fl/fl and Vil1creIl33fl/fl (G) Il1rl1fl/fl and ChgacreERIl1rl1fl/fl (H) mice were injected with PBS or IL-33. Time to cessation of bleeding in response to tail injury was assessed 10 minutes after injection. (I) Representative histograms of fluorescence intensity of CD62P and Jon/A for platelets treated with or without thrombin between Tph1fl/fl and Vil1creTph1fl/fl mice. (J) Quantification of geometric mean fluorescence intensity in panel I. (K) Tph1fl/fl and Vil1creTph1fl/fl mice were injected with PBS or IL-33. Time to cessation of bleeding in response to tail injury was assessed 10 minutes after injection. Data are representative of 2 independent experiments (A,C,E,I) or are pooled from 2 independent experiments (B,D, F-H, J-K). *P < .05, **P < .01, ***P < .001 by Student t test. Error bars represent standard deviation. NS, not statistically significant.
IL-33 rapidly enhances platelet function for hemostasis. (A-F) Indicated mice were injected with PBS or IL-33. Platelet activities were assessed by flow cytometry 10 minutes after injection. Representative histograms of fluorescence intensity and quantification of geometric mean fluorescence intensity of CD62P and Jon/A for platelets treated with or without thrombin from WT (A-B), Vil1creIl33fl/fl (C-D), and ChgacreERIl1rl1fl/fl (E-F) mice. (G-H) Il33fl/fl and Vil1creIl33fl/fl (G) Il1rl1fl/fl and ChgacreERIl1rl1fl/fl (H) mice were injected with PBS or IL-33. Time to cessation of bleeding in response to tail injury was assessed 10 minutes after injection. (I) Representative histograms of fluorescence intensity of CD62P and Jon/A for platelets treated with or without thrombin between Tph1fl/fl and Vil1creTph1fl/fl mice. (J) Quantification of geometric mean fluorescence intensity in panel I. (K) Tph1fl/fl and Vil1creTph1fl/fl mice were injected with PBS or IL-33. Time to cessation of bleeding in response to tail injury was assessed 10 minutes after injection. Data are representative of 2 independent experiments (A,C,E,I) or are pooled from 2 independent experiments (B,D, F-H, J-K). *P < .05, **P < .01, ***P < .001 by Student t test. Error bars represent standard deviation. NS, not statistically significant.
Intestinal IL-33–ST2 axis is critical for neutrophil recruitment via platelet-derived 5-HT
Loss of peripheral 5-HT leads to dampened neutrophil migration to the site of inflammation.9 Also, neutrophil recruitment is often observed during the early phase of inflammation.31 We induced acute peritonitis and found reduced retrieved neutrophils in the abdominal cavity of Vil1creIl33fl/fl mice compared with control mice (Figure 4A-C). Similarly, ChgacreERIl1rl1fl/fl mice also exhibited impaired neutrophil recruitment during acute peritonitis (Figure 4D-F), suggesting that the intestinal IL-33–ST2 axis is critical for neutrophil extravasation during acute inflammation. To further address whether IL-33–mediated neutrophil migration is 5-HT dependent, we examined Vil1creTph1fl/fl mice and found fewer abdominal neutrophils after acute peritonitis induction compared with controls (Figure 4G-I). Moreover, IL-33 injection resulted in elevated neutrophil recruitment in control mice after induction of acute peritonitis, whereas such changes were abolished in Vil1creTph1fl/fl mice (Figure 4G-I). Thus, we demonstrated that intestinal IL-33 promotes neutrophil extravasation during acute inflammation through 5-HT.
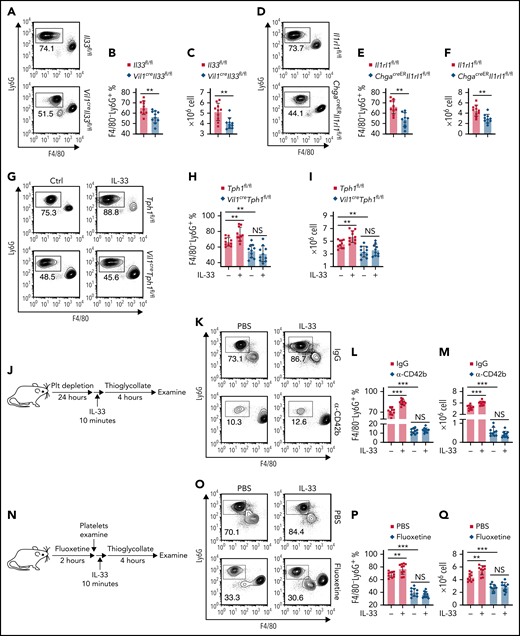
Intestinal IL-33–ST2 signaling promotes neutrophil recruitment in acute peritonitis via platelet-derived 5-HT. (A-F) Intraperitoneal injection of 4% thioglycollate into Il33fl/fl and Vil1creIl33fl/fl (A-C) or Il1rl1fl/fl and ChgacreERIl1rl1fl/fl (D-F) mice. (A,D) Flow cytometric analysis of lymphocytes isolated from peritoneum 4 hours after thioglycollate administration. (B,E) Quantification of percentile of gated F4/80–Ly6G+ neutrophils in panels A and D. (D,F) Quantification of F4/80−Ly6G+ neutrophils isolated from peritoneum 4 hours after thioglycollate administration. (G-I) Tph1fl/fl and Vil1creTph1fl/fl mice were treated with PBS or IL-33 followed by a 10-minute wait. Intraperitoneal injection of 4% thioglycollate into Tph1fl/fl and Vil1creTph1fl/fl mice. (G) Flow cytometric analysis of lymphocytes isolated from peritoneum 4 hours after thioglycollate administration with or without IL-33 treatment. (H) Quantification of percentile of gated F4/80−Ly6G+ neutrophils in panel G. (I) Quantification of F4/80−Ly6G+ neutrophils isolated from peritoneum 4 hours after thioglycollate administration with or without IL-33 treatment. (J) Schematic illustration of platelet depletion in WT mice, followed by intraperitoneal injection of 4% thioglycolate with or without IL-33 injection. (K) Flow cytometric analysis of lymphocytes isolated from peritoneum as in panel J. (L) Quantification of percentile of gated F4/80−Ly6G+ neutrophils in panel K. (M) Quantification of F4/80−Ly6G+ neutrophils isolated from peritoneum 4 hours after peritonitis induction. (N) Schematic illustration of fluoxetine treatment in WT mice, followed by intraperitoneal injection of 4% thioglycolate. (O) Flow cytometric analysis of lymphocytes isolated from peritoneum as in panel N. (P) Quantification of percentile of gated F4/80−Ly6G+ neutrophils in panel O. (Q) Quantification of F4/80−Ly6G+ neutrophils isolated from peritoneum 4 hours after peritonitis induction with or without 10-minute IL-33 treatment. Data are representative of 2 independent experiments (A, D, G, K, O) or are pooled from 2 independent experiments (B, C, E-F, H-I, L-M, P-Q). *P < .05, **P < .01, ***P < .001 by Student t test. Error bars represent standard deviation. NS, not statistically significant.
Intestinal IL-33–ST2 signaling promotes neutrophil recruitment in acute peritonitis via platelet-derived 5-HT. (A-F) Intraperitoneal injection of 4% thioglycollate into Il33fl/fl and Vil1creIl33fl/fl (A-C) or Il1rl1fl/fl and ChgacreERIl1rl1fl/fl (D-F) mice. (A,D) Flow cytometric analysis of lymphocytes isolated from peritoneum 4 hours after thioglycollate administration. (B,E) Quantification of percentile of gated F4/80–Ly6G+ neutrophils in panels A and D. (D,F) Quantification of F4/80−Ly6G+ neutrophils isolated from peritoneum 4 hours after thioglycollate administration. (G-I) Tph1fl/fl and Vil1creTph1fl/fl mice were treated with PBS or IL-33 followed by a 10-minute wait. Intraperitoneal injection of 4% thioglycollate into Tph1fl/fl and Vil1creTph1fl/fl mice. (G) Flow cytometric analysis of lymphocytes isolated from peritoneum 4 hours after thioglycollate administration with or without IL-33 treatment. (H) Quantification of percentile of gated F4/80−Ly6G+ neutrophils in panel G. (I) Quantification of F4/80−Ly6G+ neutrophils isolated from peritoneum 4 hours after thioglycollate administration with or without IL-33 treatment. (J) Schematic illustration of platelet depletion in WT mice, followed by intraperitoneal injection of 4% thioglycolate with or without IL-33 injection. (K) Flow cytometric analysis of lymphocytes isolated from peritoneum as in panel J. (L) Quantification of percentile of gated F4/80−Ly6G+ neutrophils in panel K. (M) Quantification of F4/80−Ly6G+ neutrophils isolated from peritoneum 4 hours after peritonitis induction. (N) Schematic illustration of fluoxetine treatment in WT mice, followed by intraperitoneal injection of 4% thioglycolate. (O) Flow cytometric analysis of lymphocytes isolated from peritoneum as in panel N. (P) Quantification of percentile of gated F4/80−Ly6G+ neutrophils in panel O. (Q) Quantification of F4/80−Ly6G+ neutrophils isolated from peritoneum 4 hours after peritonitis induction with or without 10-minute IL-33 treatment. Data are representative of 2 independent experiments (A, D, G, K, O) or are pooled from 2 independent experiments (B, C, E-F, H-I, L-M, P-Q). *P < .05, **P < .01, ***P < .001 by Student t test. Error bars represent standard deviation. NS, not statistically significant.
To further address the link between neutrophil extravasation and IL-33–mediated platelet activation, we depleted the platelets in WT mice32 (Figure 4J) and found that mice with depleted platelets exhibited reduced neutrophil extravasation compared with rat immunoglobulin G–treated mice during peritonitis (Figure 4K-M). Furthermore, IL-33 treatment failed in promoting peritoneal neutrophil recruitment in platelet-depleted mice (Figure 4K-M). Next, we treated mice with fluoxetine, the selective 5-HT reuptake inhibitor, to pharmacologically deplete platelet 5-HT33 (Figure 4N). Fluoxetine treatment resulted in impaired platelet activation without affecting peripheral 5-HT levels (supplemental Figure 3C-E). Importantly, IL-33 injection induced fewer peritoneal neutrophils in fluoxetine-treated mice compared with control mice (Figure 4O-Q). These results indicate that 5-HT–mediated platelet activation is critical for IL-33–induced neutrophil recruitment. Lastly, we restored 5-HT in Il33−/− mice through 5-HTP in the drinking water34 (supplemental Figure 3F). We found elevated platelet activation (supplemental Figure 3G-I) and hemostasis (supplemental Figure 3J) in 5-HTP–treated mice, indicating that supplementation with 5-HT enhances platelet activity in Il33−/− mice. Moreover, we depleted platelets before peritonitis induction and examined neutrophil infiltration 4 hours after peritonitis induction (supplemental Figure 3G). We found that loss of platelets abolished the differences in neutrophil recruitment between 5-HTP supplementation and PBS, indicating that 5-HT–mediated neutrophil extravasation is platelet dependent (supplemental Figure 3K-M). Together, these data provide a direct link between IL-33–mediated platelet activation and neutrophil extravasation.
Intestinal IL-33–ST2 axis is critical for wound healing
By recruiting neutrophils, platelet 5-HT also influences wound healing.9 We examined neutrophil extravasation after inducing aseptic skin wounds. We noticed that Vil1creTph1fl/fl mice exhibited less neutrophil recruitment to the site of injury compared with control mice by histologic analysis and neutrophil enzyme MPO assessment (Figure 5A-B). Also, IL-33 administration promoted neutrophil levels in the wound sites of control mice but not in Vil1creTph1fl/fl mice (Figure 5A-B), indicating that IL-33–induced neutrophil recruitment during acute skin injury is mediated by 5-HT. Next, we found that loss of IL-33 in the gut resulted in less neutrophil recruitment to the site of injury (Figure 5C-D), leading to a prolonged wound closure process in Vil1creIl33fl/fl mice (Figure 5E-F). It has been reported that IL-33 promotes skin wound healing via regulating the function of local immune responses.35-37 We detected no change of local IL-33 at wound sites between Vil1creIl33fl/fl and controls (supplemental Figure 4A), suggesting that gut-derived IL-33 does not affect local IL-33 levels at remote injury sites but instead promotes EC cell–derived 5-HT release to influence skin inflammation. Similarly, ChgacreERIl1rl1fl/fl mice exhibited impaired neutrophil recruitment during skin injury (Figure 5G-H), accompanied with delayed wound healing without alteration in local IL-33 levels (Figure 5I-J; supplemental Figure 4B). Lastly, although loss of the intestinal IL-33–ST2 axis impaired platelet activation, the same levels of platelet recruitment were observed at the injury sites between Vil1creIl33fl/fl, ChgacreERIl1rl1fl/fl, and control mice, suggesting that the defect in neutrophil levels in the wounds was a result of impaired platelet activation but not recruitment in the absence of IL-33 signaling (supplemental Figure 4C-D). Together, our data indicate the important role of 5-HT release induced by the intestinal IL-33–ST2 axis in neutrophil recruitment during acute inflammation.
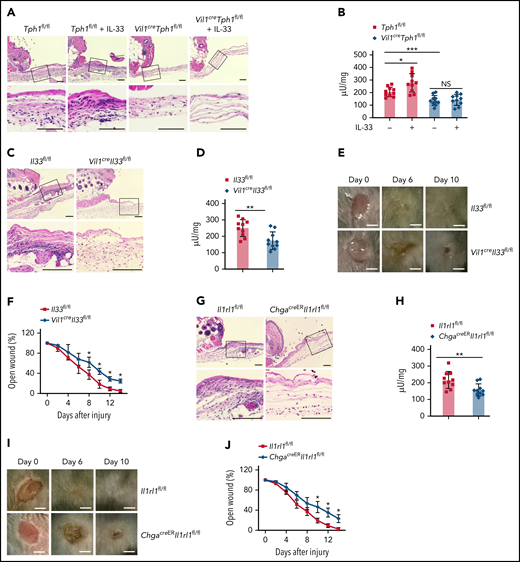
Intestinal IL-33–ST2 signaling regulates axis neutrophil recruitment during acute skin injury via peripheral 5-HT. (A-B) Aseptic skin wounds were punched into the dorsal skin on Tph1fl/fl and Vil1creTph1fl/fl mice with or without IL-33 treatment. (A) Wounded skin samples were collected 4 hours after injury and processed by hematoxylin and eosin (HE) staining. Inset: magnified skin tissue at the injury site. Scale bar, 100 μm. (B) Tissue neutrophil enzyme MPO was assessed from the wound sites. (C-J) Aseptic skin wounds were punched into the dorsal skin on Il33fl/fl and Vil1creIl33fl/fl mice (C-F) or Il1rl1fl/fl and ChgacreERIl1rl1fl/fl mice (G-J). (C,G) Wounded skin samples were collected 4 hours after injury and processed by HE staining. Inset: magnified skin tissue at the injury site. Scale bar, 100 μm. (D,H) Tissue neutrophil enzyme MPO was assessed from the wound sites. (E,I) The Wound sites were photographed at the indicated time. Day-0 picture was taken immediately after the injury. Scale bar, 2.5 mm. (F,J) Graph showing wound closure rate. Data are representative of 2 independent experiments (A, C, E, G, I) or are pooled from 2 independent experiments (B, D, F, H, J). *P < .05, **P < .01 by Student t test. Error bars represent standard deviation. NS, not statistically significant.
Intestinal IL-33–ST2 signaling regulates axis neutrophil recruitment during acute skin injury via peripheral 5-HT. (A-B) Aseptic skin wounds were punched into the dorsal skin on Tph1fl/fl and Vil1creTph1fl/fl mice with or without IL-33 treatment. (A) Wounded skin samples were collected 4 hours after injury and processed by hematoxylin and eosin (HE) staining. Inset: magnified skin tissue at the injury site. Scale bar, 100 μm. (B) Tissue neutrophil enzyme MPO was assessed from the wound sites. (C-J) Aseptic skin wounds were punched into the dorsal skin on Il33fl/fl and Vil1creIl33fl/fl mice (C-F) or Il1rl1fl/fl and ChgacreERIl1rl1fl/fl mice (G-J). (C,G) Wounded skin samples were collected 4 hours after injury and processed by HE staining. Inset: magnified skin tissue at the injury site. Scale bar, 100 μm. (D,H) Tissue neutrophil enzyme MPO was assessed from the wound sites. (E,I) The Wound sites were photographed at the indicated time. Day-0 picture was taken immediately after the injury. Scale bar, 2.5 mm. (F,J) Graph showing wound closure rate. Data are representative of 2 independent experiments (A, C, E, G, I) or are pooled from 2 independent experiments (B, D, F, H, J). *P < .05, **P < .01 by Student t test. Error bars represent standard deviation. NS, not statistically significant.
Loss of intestinal IL-33 improves survival in LPS endotoxic shock
It is known that during septic shock, neutrophil-induced acute inflammation contributes to host mortality.38 Plus, peripheral 5-HT has been shown to lower survival in LPS endotoxic shock.9 Considering IL-33 promotes 5-HT release,29 we examined the effect of IL-33 on sepsis. We administrated E coli serotype 055:B5 LPS and observed significantly reduced mortality in Vil1creTph1fl/fl mice compared with controls (Figure 6A). Tph1fl/fl control mice exhibited worse survival rates with pretreatment of IL-33, whereas such an effect of IL-33 was diminished in Vil1creTph1fl/fl mice (Figure 6A). Consistently, Vil1creTph1fl/fl mice exhibited abolished IL-33–induced neutrophil migration into the peritoneum compared with Tph1fl/fl mice after LPS administration (Figure 6B-D). Moreover, lower levels of interferon-γ, tumor necrosis factor-α, and IL-6 were noticed in Vil1creTph1fl/fl mice after LPS injection (Figure 6E; supplemental Figure 5A). Vil1creTph1fl/fl mice were also protected from LPS-induced hypothermia (supplemental Figure 5B; Figure 6F). Importantly, IL-33 injection led to increased levels of proinflammatory cytokines and lowered body temperatures in control mice but not in Vil1creTph1fl/fl mice (Figure 6E-F; supplemental Figure 5A). These results indicate that IL-33 exacerbates neutrophil recruitment and endotoxic shock by promoting peripheral 5-HT levels.
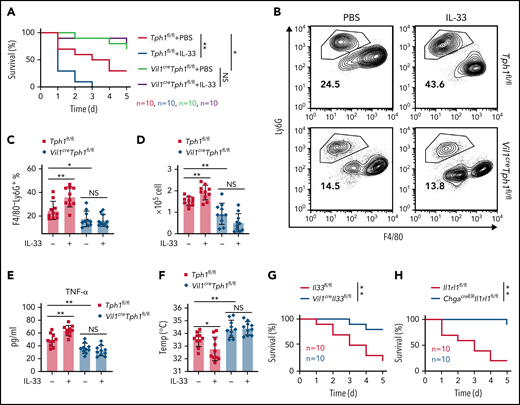
IL-33 reduces survival of LPS endotoxic shock via peripheral 5-HT. (A-F) Intraperitoneal injection of E coli serotype 055:B5 LPS into Tph1fl/fl and Vil1creTph1fl/fl mice with or without IL-33 treatment. (A) Survival rate after LPS injection. (B) Flow cytometric analysis of lymphocytes isolated from peritoneum 4 hours after LPS injection. (C) Quantification of percentile of gated F4/80−Ly6G+ neutrophils in panel B. (D) Quantification of F4/80−Ly6G+ neutrophils isolated from peritoneum 4 hours after LPS injection. (E) Tumor necrosis factor-α (TNF-α) levels in the serum at 24 hours after LPS injection. (F) Body temperature was measured at 4 hours after LPS injection. (G-H) Intraperitoneal injection of E coli serotype 055:B5 LPS into Il33fl/fl and Vil1creIl33fl/fl mice (G) or Il1rl1fl/fl and ChgacreERIl1rl1fl/fl mice (H). Survival rate after LPS injection was monitored. Data are representative of 2 independent experiments (B) or are pooled from 2 independent experiments (A, C-H). *P < .05, **P < .01, ***P < .001 by Student t test. Error bars represent standard deviation. IFN-γ, interferon-γ; NS, not statistically significant.
IL-33 reduces survival of LPS endotoxic shock via peripheral 5-HT. (A-F) Intraperitoneal injection of E coli serotype 055:B5 LPS into Tph1fl/fl and Vil1creTph1fl/fl mice with or without IL-33 treatment. (A) Survival rate after LPS injection. (B) Flow cytometric analysis of lymphocytes isolated from peritoneum 4 hours after LPS injection. (C) Quantification of percentile of gated F4/80−Ly6G+ neutrophils in panel B. (D) Quantification of F4/80−Ly6G+ neutrophils isolated from peritoneum 4 hours after LPS injection. (E) Tumor necrosis factor-α (TNF-α) levels in the serum at 24 hours after LPS injection. (F) Body temperature was measured at 4 hours after LPS injection. (G-H) Intraperitoneal injection of E coli serotype 055:B5 LPS into Il33fl/fl and Vil1creIl33fl/fl mice (G) or Il1rl1fl/fl and ChgacreERIl1rl1fl/fl mice (H). Survival rate after LPS injection was monitored. Data are representative of 2 independent experiments (B) or are pooled from 2 independent experiments (A, C-H). *P < .05, **P < .01, ***P < .001 by Student t test. Error bars represent standard deviation. IFN-γ, interferon-γ; NS, not statistically significant.
Next, given the reduced peripheral 5-HT in Vil1creIl33fl/fl and ChgacreERIl1rl1fl/fl mice, we hypothesized that loss of intestinal IL-33 signaling could protect mice from septic shock. Indeed, significantly reduced mortality was seen in Vil1creIl33fl/fl compared with control mice (Figure 6G), associated with reduced peritoneal neutrophil recruitment (supplemental Figure 5C-E), reduced inflammatory cytokines (supplemental Figure 5F), and enhanced body temperatures (supplemental Figure 5G) after LPS administration. Similar phenotypes were also observed in ChgacreERIl1rl1fl/fl mice during endotoxic shock (Figure 6H; supplemental Figure 5H-L). These data suggest that intestinal IL-33–ST2 signaling contributes to the pathogenesis of systemic septic shock.
Neutralization of IL-33 reduces peripheral 5-HT and protects mice from LPS endotoxic shock
To further investigate the therapeutic potential in disorders mediated by IL-33–induced 5-HT, we applied soluble ST2 (sST2) to neutralize extracellular IL-33.39,40 Injection of the sST2-Fc–expressing plasmid41 resulted in reduced serum 5-HT levels and platelet activity in Tph1fl/fl but not Vil1creTph1fl/fl mice (Figure 7A; supplemental Figure 6A-B). Consequently, we found sST2 treatment prolonged bleeding time without changing platelet counts, whereas such an effect was abolished in Vil1creTph1fl/fl mice (supplemental Figure 6C-D), suggesting that neutralization of IL-33 blocks 5-HT release, repressing platelet activation. Next, we pretreated mice with sST2 and noticed that Tph1fl/fl mice exhibited elevated survival rates during LPS endotoxic shock, whereas no effect of sST2 was seen in Vil1creTph1fl/fl mice (Figure 7B). Consistently, fewer neutrophils, reduced proinflammatory cytokines, and higher body temperatures were found in sST2-pretreated control mice after LPS administration, but these changes were not seen in Vil1creTph1fl/fl mice with sST2 treatment (Figure 7C-G; supplemental Figure 6E). Together, these data indicate that sST2 treatment improves the survival rate in septic shock via suppressing 5-HT–mediated excessive neutrophil extravasation.
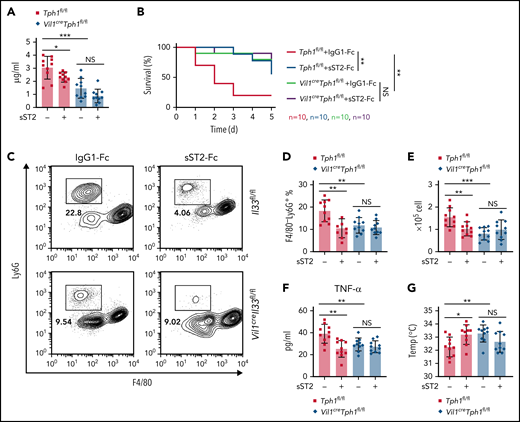
Neutralization of IL-33 reduces peripheral 5-HT and protects mice from LPS endotoxic shock. (A) Control IgG-Fc– or sST2-Fc–expressing plasmid was IV injected into Tph1fl/fl and Vil1creTph1fl/fl mice. Relative 5-HT amounts in serum were assessed by ELISA. (B-G) IP injection of E coli serotype 055:B5 LPS into Tph1fl/fl and Vil1creTph1fl/fl mice with 1-day pretreatment of IgG-Fc– or sST2-Fc–expressing plasmid. (B) Survival rate after LPS injection. (C) Flow cytometric analysis of lymphocytes isolated from peritoneum. (D) Quantification of frequency of gated F4/80−Ly6G+ neutrophils in panel C. (E) Quantification of F4/80−Ly6G+ neutrophils isolated from peritoneum 4 hours after LPS injection. (F) Tumor necrosis factor-α (TNF-α) levels in the serum at 24 hours after LPS injection. (G) Body temperature was measured at 0 and 4 hours after LPS injection. Data are representative of 2 independent experiments (C) or are pooled from 2 independent experiments (A-B, D-G). *P < .05, **P < .01, ***P < .001 by Student t test. Error bars represent standard deviation. NS, not statistically significant.
Neutralization of IL-33 reduces peripheral 5-HT and protects mice from LPS endotoxic shock. (A) Control IgG-Fc– or sST2-Fc–expressing plasmid was IV injected into Tph1fl/fl and Vil1creTph1fl/fl mice. Relative 5-HT amounts in serum were assessed by ELISA. (B-G) IP injection of E coli serotype 055:B5 LPS into Tph1fl/fl and Vil1creTph1fl/fl mice with 1-day pretreatment of IgG-Fc– or sST2-Fc–expressing plasmid. (B) Survival rate after LPS injection. (C) Flow cytometric analysis of lymphocytes isolated from peritoneum. (D) Quantification of frequency of gated F4/80−Ly6G+ neutrophils in panel C. (E) Quantification of F4/80−Ly6G+ neutrophils isolated from peritoneum 4 hours after LPS injection. (F) Tumor necrosis factor-α (TNF-α) levels in the serum at 24 hours after LPS injection. (G) Body temperature was measured at 0 and 4 hours after LPS injection. Data are representative of 2 independent experiments (C) or are pooled from 2 independent experiments (A-B, D-G). *P < .05, **P < .01, ***P < .001 by Student t test. Error bars represent standard deviation. NS, not statistically significant.
Discussion
IL-33 was found to promote neutrophil migration via induction of macrophage-derived chemokines and cytokines, which act on neutrophils during autoimmunity. The same study suggests that IL-33 can directly act as a chemoattractant for neutrophils.42 Based on our previous findings that IL-33 induces intestinal 5-HT release,29 we show here that IL-33–mediated 5-HT promotes neutrophil extravasation via activation of platelets. Clinical studies indicate that neutropenic individuals often have difficulty in wound healing,43,44 suggesting the function of neutrophils in promoting would healing. Other mouse studies have also indicated that reduced neutrophil recruitment to wounds results in delayed healing.45,46 More importantly, 5-HT–mediated platelet activation has been shown to promote neutrophil recruitment during wound healing, accelerating wound closure,9 which is consistent with our findings. Thus, the role of neutrophils could be various during cutaneous wound healing, probably depending on different host conditions and inflammatory milieu.
Although it has been extensively studied, the precise role of the IL-33–ST2 axis in septic shock is still unclear, depending on the experimental models used in studies. For instance, IL-33 has been found to elevate the production of proinflammatory cytokines from macrophages after LPS administration,47 which is consistent with our data. However, in a mouse model of cecal ligation and puncture–induced sepsis, exogenous IL-33 promotes neutrophil recruitment by preserving CXCR2 expression on neutrophils, resulting in better bacterial clearance and less mortality.48,49 Considering our findings in wound healing and sepsis with IL-33 treatment, we conclude that IL-33 could regulate neutrophil recruitment through both 5-HT–dependent and –independent manners. The discrepancy in survival rates between LPS- and cecal ligation and puncture–induced sepsis after IL-33 treatment is possibly due to whether recruited neutrophils induce excessive inflammation or bacterial clearance. On the other hand, it has been reported that ST2 signaling promotes endotoxin tolerance. ST2-deficient mice exhibit greater susceptibility to LPS-induced sepsis.50 In those studies, germ line ST2-deficient mice were used, and the role of ST2 in sepsis was focused on macrophage activation.47,50 By deleting ST2 specifically on EC cells, our data reveal a new role of ST2 in enhancing sensitivity in septic shock through 5-HT release. Therefore, the conflicting outcomes of sepsis between Il1rl1−/− and ChgacreERIl1rl1fl/fl mice indicate distinct roles of ST2 in different cell types during LPS-mediated pathogenesis.
The functions of sST2 have been tested in different studies, given the critical role of IL-33 in LPS-mediated proinflammatory responses.51-53 In line with our results, sST2 treatment decreases LPS-induced proinflammatory cytokine production by macrophages and thus leads to improved survival rates in endotoxic shock.52 Meanwhile, it is known that 5-HT–mediated neutrophil extravasation triggers an inflammatory cascade of cytokine production during septic shock.9 Although we show that sST2 treatment results in reduction of both peripheral 5-HT and proinflammatory cytokines after LPS injection, data from Vil1creTph1fl/fl mice indicate that sST2 plays a protective role in sepsis, primarily via 5-HT. Thus, our work demonstrates that sST2 exerts its anti-inflammatory function by not only downregulating cytokine transcription but also inhibiting 5-HT release. Our findings also provide therapeutic insights into platelet-associated inflammation and tissue damage. It is known that patients with sepsis exhibit dramatic reductions of 5-HT content in platelets.54 Amelioration of excessive neutrophil extravasation by IL-33 neutralization could thus be a potential antiserotonergic therapeutic approach for sepsis, although potential complications of hemorrhage should be taken into consideration.
In summary, we show here that the intestinal IL-33–ST2 axis influences systemic platelet function through enhancing 5-HT release. We show that intestinal IL-33–mediated platelet activation promotes neutrophil recruitment during acute inflammation, revealing the distal regulatory function of gut-derived IL-33. Thus, our data indicate the significance of the distinct function of IL-33 at different barrier sites, which could effectively modulate local environments and subsequently affect systemic physiology.
Acknowledgments
The authors thank Susumu Nakae (University of Tokyo, Tokyo, Japan) for Il33−/− mice, Gerard Karsenty (Columbia University) for Tph1flox mice, Andrew B. Leiter (University of Massachusetts Medical School) for Tph1CFP mice, Giorgio Trinchieri (National Cancer Institute) for the Il1rl1−/− mice, and Keizo Takenaga (Shimane University Faculty of Medicine) for sST2-Fc–expressing plasmids.
This work was supported by National Multiple Sclerosis Society Career Transition Award TA 3059-A-2 to C.W.
Authorship
Contribution: Z.C. and C.W. conceived the project; Z.C., J. Luo, J. Li, G.K., A.S., and Y.H. performed experiments and data analysis; Z.C., J. Luo, Y.H., and C.W. wrote and edited the manuscript; and C.W. supervised the project.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Chuan Wu, National Cancer Institute, 9000 Rockville Pike, Bldg 10, Rm 4B17, Bethesda, MD 20892; e-mail: chuan.wu@nih.gov.
Requests for data sharing may be submitted to Chuan Wu (chuan.wu@nih.gov).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal