

Key Points
Immune-compatible donor memory-like NK cells expand and functionally persist for months after a single dose without exogenous cytokines.
Combined with standard therapy, donor ML NK cells safely induce remissions in poor prognosis, pediatric, relapsed AML after HCT.

Abstract
Pediatric and young adult (YA) patients with acute myeloid leukemia (AML) who relapse after allogeneic hematopoietic cell transplantation (HCT) have an extremely poor prognosis. Standard salvage chemotherapy and donor lymphocyte infusions (DLIs) have little curative potential. Previous studies showed that natural killer (NK) cells can be stimulated ex vivo with interleukin-12 (IL-12), -15, and -18 to generate memory-like (ML) NK cells with enhanced antileukemia responses. We treated 9 pediatric/YA patients with post-HCT relapsed AML with donor ML NK cells in a phase 1 trial. Patients received fludarabine, cytarabine, and filgrastim followed 2 weeks later by infusion of donor lymphocytes and ML NK cells from the original HCT donor. ML NK cells were successfully generated from haploidentical and matched-related and -unrelated donors. After infusion, donor-derived ML NK cells expanded and maintained an ML multidimensional mass cytometry phenotype for >3 months. Furthermore, ML NK cells exhibited persistent functional responses as evidenced by leukemia-triggered interferon-γ production. After DLI and ML NK cell adoptive transfer, 4 of 8 evaluable patients achieved complete remission at day 28. Two patients maintained a durable remission for >3 months, with 1 patient in remission for >2 years. No significant toxicity was experienced. This study demonstrates that, in a compatible post-HCT immune environment, donor ML NK cells robustly expand and persist with potent antileukemic activity in the absence of exogenous cytokines. ML NK cells in combination with DLI present a novel immunotherapy platform for AML that has relapsed after allogeneic HCT. This trial was registered at https://clinicaltrials.gov as #NCT03068819.
Introduction
AML comprises 18% of all leukemia diagnoses in children and young adults (YAs).1 Treatment outcomes for AML have improved based on optimized chemotherapy regimens and risk stratification, with overall survival reaching ∼70%.2-5 Standard treatment centers on high-dose chemotherapy using anthracyclines and cytarabine, which results in complete remission (CR) in 80% to 90% of patients, followed by consolidation therapy with additional chemotherapy (low risk) or allogeneic hematopoietic cell transplantation (HCT, high risk).4,6,7 Despite this high remission rate, ∼40% of patients relapse.4,6-9 Outcomes for relapsed AML remain extremely poor, despite significant efforts using intensified reinduction chemotherapy and HCT in second remission.9-14 AML that relapses after HCT presents further challenges, as it is often refractory to chemotherapy. Donor lymphocyte infusions (DLIs) have been used to boost graft-versus-leukemia effect; however, DLIs have limited efficacy, with <11% of patients achieving remission.15-17 Approaches using salvage chemotherapy followed by a second HCT have had limited success and high toxicity.9-14 New therapies with favorable toxicity profiles are needed for pediatric and YA patients with AML that has relapsed after HCT.
Recent clinical successes with chimeric antigen receptor T cells in pediatric acute lymphoblastic leukemia highlighted the promise of cellular immunotherapy.18-20 However, developing T-cell–based immunotherapy for AML has been hindered by lack of suitable antigenic targets.21,22 An alternative approach using natural killer (NK) cells for AML therapy has shown promise.23-26 NK cells are cytotoxic innate lymphoid cells specialized to eliminate virus-infected and malignantly transformed target cells.27,28 In contrast to T cells, they recognize AML cells by integrating signals received through germline DNA-encoded activating and inhibitory receptors that sense stress-induced ligands and HLA. Clinical studies for patients with AML who undergo haploidentical allogeneic HCT have shown that donor NK cell reactivity against the leukemia predicts long-term disease-free survival.29,30 NK cell receptor studies further support a role for NK cells in graft-versus-leukemia responses.31-33 Enriched, allogeneic NK cell products adoptively transferred into patients have been shown to be safe and have induced short remissions in patients with active AML.34-38 Overall, these prior studies have had mixed results with limited donor NK cell persistence. Enhancing the persistence and antileukemia activity of donor NK cells is needed to advance the therapeutic potential of adoptive NK cell immunotherapy.39-41
In previous studies, we have shown that human NK cells stimulated ex vivo with interleukin (IL)-12, -15, and -18 differentiate into memory-like (ML) NK cells with enhanced antileukemia activity.42 These ML NK cells show enhanced proliferation, expression of the high-affinity IL-2 receptor (IL-2Rαβγ, CD25), increased cytotoxicity against leukemia targets, and increased interferon-γ (IFN-γ) production after stimulation.42-47 Furthermore, a phase 1 clinical trial of adoptive haploidentical ML NK cellular therapy in adults with relapsed/refractory AML safely induced remission in 47% of patients.46,47 Infused haploidentical donor ML NK cells expanded and persisted in vivo, but were eliminated after recovery of recipient T cells, thereby providing a 2- to 3-week window of therapeutic opportunity.46,47 However, the expansion and persistence potential of ML NK cells remains unclear.
To address this gap in our understanding, we hypothesized that providing ML NK cells that are matched to the recipient’s T cells would avoid allorejection and provide a longer opportunity to persist and function. In addition, preclinical data showed ML NK cells can use T-cell–derived IL-2 to enhance their function and persistence, replacing the need for exogenous cytokines.44,48,49 To test these concepts, we designed a phase 1 clinical trial combining standard-of-care therapy (salvage chemotherapy with a T-cell based DLI) with donor ML NK cells to treat pediatric/YA AML that relapsed after HCT. Patients received fludarabine and cytarabine for leukemic reduction followed by infusion of a DLI and ML NK cell product generated from the same donor as the original HCT. We report on ML NK cell expansion, persistence, and function after administration in this new immunologic situation, as well as the safety and antileukemia activity of this previously unreported combination immunotherapy.
Materials and methods
Study design
All patients aged 1 to 30 years treated in a phase 1 clinical trial between November 2018 and May 2020 are included. Written, informed consent was obtained for all patients and donors. The study was approved by the Washington University School of Medicine Institutional Review Board (IRB#201709041). Patients were treated with fludarabine (30 mg/m2 daily), cytarabine (2000 mg/m2 daily), and granulocyte colony-stimulating factor (filgrastim, 5 μg/kg daily; maximum dose, 300 μg) for 5 days, followed 2 to 4 weeks later with infusion of donor T-cell-based DLI and donor ML NK cells (Figure 1). To generate the products, a nonmobilized leukapheresis product was collected from the HCT donor and processed into DLI and ML NK cells. DLI (1 × 106 T cells per kg) was immediately infused into the patient. NK cells were purified from the remaining product by CD3 depletion, followed by CD56+ selection on a CliniMACS device (mean purity, 94% ± 1% CD3−CD56+). Purified NK cells were stimulated with IL-12, -15, and -18 for 12 to 16 hours under current good manufacturing practice conditions to generate ML NK cells, as described.46,47 NK cells were washed and infused into patients (maximum dose, 10 × 106 cells per kg). Patients were routinely monitored for clinical response, graft-versus-host disease (GVHD), and adverse events. Clinical responses were defined by International Working Group criteria.50 Adverse events were defined using Common Terminology Criteria for Adverse Events (CTCAE), v4.0. Correlative samples were obtained from peripheral blood and bone marrow at defined times after NK cell infusion. Additional details are available in the supplemental Appendix, available on the Blood Web site. This interim analysis was completed to assess safety and clinical responses before modifications to extend enrollment and include a second course of therapy.
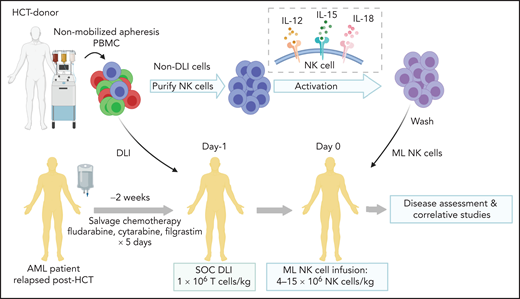
Treatment schema for clinical trial combining salvage chemotherapy, DLI, and ML NK cell infusion. Patients provided consent and were enrolled in clinical trial NCT03068819. Salvage chemotherapy with fludarabine, cytarabine, and granulocyte colony stimulating factor was administered 2 to 4 weeks before ML NK cell infusion. On day −1, a nonmobilized leukapheresis product was collected from the same donor as HCT. T-cell DLI (1 × 106 T cells per kg) was immediately infused into the patient. The remainder of the apheresis product was enriched for NK cells, which were then stimulated overnight with IL-12, L-15, and -18 to generate ML NK cells, which were infused into the patient the next day (day 0; max dose, 10 × 106 cells per kg). Disease assessment (clinical response) and adverse events were subsequently monitored, and samples for were collected for correlative experiments. SOC, standard of care.
Treatment schema for clinical trial combining salvage chemotherapy, DLI, and ML NK cell infusion. Patients provided consent and were enrolled in clinical trial NCT03068819. Salvage chemotherapy with fludarabine, cytarabine, and granulocyte colony stimulating factor was administered 2 to 4 weeks before ML NK cell infusion. On day −1, a nonmobilized leukapheresis product was collected from the same donor as HCT. T-cell DLI (1 × 106 T cells per kg) was immediately infused into the patient. The remainder of the apheresis product was enriched for NK cells, which were then stimulated overnight with IL-12, L-15, and -18 to generate ML NK cells, which were infused into the patient the next day (day 0; max dose, 10 × 106 cells per kg). Disease assessment (clinical response) and adverse events were subsequently monitored, and samples for were collected for correlative experiments. SOC, standard of care.
Flow cytometry
Flow cytometric analyses were performed with previously described staining, acquisition, and analysis methods.46,47
Mass cytometry
Lymphocytes were processed fresh or from cryopreserved samples for mass cytometry, as previously described and detailed in supplemental Methods.47 Lymphocyte subsets were identified using FlowSOM (supplemental Table 1), and metaclusters were then manually annotated, based on expression of lineage markers. The metacluster identified as NK cells was used for subsequent phenotypic analysis.47 Peripheral blood samples ([patients] P-ML002, P-ML003, P-ML005, and P-ML009) and bone marrow samples (P-ML002, P-ML003, P-ML005, P-ML008, P-ML009, and P-ML011) were assessed. Further details on the analysis are in supplemental Methods.
Single cell RNA-sequencing
Blood samples (P-ML002 and P-ML008) were thawed in batches, then flow sorted to enrich for live NK cells by negative selection (supplemental Table 2) on a BD Aria Ilu SORP. Enriched NK cells were washed with 0.04% bovine serum albumin in phosphate-buffered saline or stained with antibodies, as previously described, omitting dextran sulfate.51 Using the 10× Chromium Instrument (5′v2 chemistry) individual cells were barcoded, and cDNA libraries were constructed and then sequenced on an Illumina NovaSeq6000. Further details on the analysis are in supplemental Methods.
Functional assays to assess cytokine production
Freshly isolated peripheral blood NK cells were restimulated with K562 cells (5:1 effector:target ratio) as previously described.46,47 Samples from P-ML002, P-ML003, P-ML005, and P-ML007 were included. The remaining patients were not assessed because of restrictions in place during COVID-19. Cells were stimulated with 1 ng/mL recombinant human IL-15. CD107a was included at the beginning of the assay, followed 1 hour later by addition of brefeldin A and monensin (GolgiPlug/GolgiStop; BD) then staining for NK-cell surface markers and intracellular IFN-γ.
Statistical analysis
Data were analyzed using Prism v9.2.0 unless otherwise noted. All data were first assessed for normal distribution. Appropriate parametric or nonparametric tests were subsequently used for statistical comparison. Methods for statistical comparisons are noted in figure legends. All comparisons used 2-sided P < .05 for significance.
Results
Patient characteristics
Ten patients were enrolled on the study. All had leukemia that had relapsed after allogeneic HCT. One patient died before starting study therapy and that patient’s data were excluded from the analyses. The remaining 9 patients ranged in age from 1.5 to 28 years (Table 1). One patient (P-ML003) had a phenotypic change from AML to T-cell lymphoblastic leukemia at the beginning of the study therapy and was observed for correlative studies but was not evaluable for efficacy assessment. All other patients had AML with high-risk characteristics. All patients received myeloablative conditioning therapy for HCT. Matched sibling, matched-unrelated, and haploidentical donors were used. Two patients completed 2 transplants before enrollment. Before ML NK cell therapy, patients received an average of 2 salvage therapies after the most recent HCT. At enrollment, bone marrow demonstrated 4% to 82% (mean, 37%) leukemic blasts. All patients had donor engraftment at enrollment.
Patient characteristics at enrollment
| P-ML ID | Age, y | Sex | Cytogenetic abnormalities | HCT donor* | HCT KIR (GvH) | HCT KIR (HvG) | Time to relapse after HCT | Post-HCT salvage therapies, n | T-cell donor engraftment, % | Blasts in BM at enrollment, % |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 1.5 | M | t:RUNX1 | Haplo | Match | Match | 90 d | 2 | 100 | 4 |
| 3 | 28 | F | Tetraploidy | Haplo | Mismatch | Match | 1 y | 0 | NA† | 70 |
| 4 | 7 | M | NUP98, FLT3-ITD | Haplo | Match | Mismatch | 60 d | 1 | 90 | 45 |
| 5 | 21 | F | −5q, MECOM | MUD | Match | Match | 150 d | 4 | NA† | 10 |
| 7 | 9 | F | −5q | MSD | Match | Match | 3 y | 3 | 95 | 62 |
| 8 | 2 | F | MECOM | MSD | Match | Match | 100 d | 2 | 77 | 11 |
| 9 | 11 | F | T(11:17) | MUD/haplo | Match | Mismatch | 160 d After HCT 2 | 2 | 100 | 15 |
| 10 | 1.5 | F | KMT2A:MLLT3 | Haplo | Mismatch | Match | 60 d | 1 | 100 | 82 |
| 11 | 4 | M | NUP98 | MUD/MUD | Match | Match | 100 d after HCT 2 | 4 | 100 | 35 |
| Average (range) | 9.4 (1.5-22) | 3 M 6 F | — | 5× Haplo 2× MUD 2× MSD | — | — | 242 d after HCT (60-1095 d) | 2 (0-4) | 96 (77-100) | 37 (4-82) |
| P-ML ID | Age, y | Sex | Cytogenetic abnormalities | HCT donor* | HCT KIR (GvH) | HCT KIR (HvG) | Time to relapse after HCT | Post-HCT salvage therapies, n | T-cell donor engraftment, % | Blasts in BM at enrollment, % |
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 1.5 | M | t:RUNX1 | Haplo | Match | Match | 90 d | 2 | 100 | 4 |
| 3 | 28 | F | Tetraploidy | Haplo | Mismatch | Match | 1 y | 0 | NA† | 70 |
| 4 | 7 | M | NUP98, FLT3-ITD | Haplo | Match | Mismatch | 60 d | 1 | 90 | 45 |
| 5 | 21 | F | −5q, MECOM | MUD | Match | Match | 150 d | 4 | NA† | 10 |
| 7 | 9 | F | −5q | MSD | Match | Match | 3 y | 3 | 95 | 62 |
| 8 | 2 | F | MECOM | MSD | Match | Match | 100 d | 2 | 77 | 11 |
| 9 | 11 | F | T(11:17) | MUD/haplo | Match | Mismatch | 160 d After HCT 2 | 2 | 100 | 15 |
| 10 | 1.5 | F | KMT2A:MLLT3 | Haplo | Mismatch | Match | 60 d | 1 | 100 | 82 |
| 11 | 4 | M | NUP98 | MUD/MUD | Match | Match | 100 d after HCT 2 | 4 | 100 | 35 |
| Average (range) | 9.4 (1.5-22) | 3 M 6 F | — | 5× Haplo 2× MUD 2× MSD | — | — | 242 d after HCT (60-1095 d) | 2 (0-4) | 96 (77-100) | 37 (4-82) |
Included are baseline characteristics, transplant history, and prior treatment history of all patients enrolled on the clinical trial who received ML NK cell immunotherapy.
F, female; GvH, graft-versus-host; haplo, haploidentical; HCT, hematopoietic cell transplant; HvG, host-versus-graft; M, male; MSD, matched sibling donor; MUD, matched unrelated donor; NA, not available.
For patients who underwent 2 transplants before enrollment, donors are list as 1st HCT/2nd HCT.
T-cell engraftment was not available for these 2 patients. Both had bone marrow tested for donor engraftment. P-ML003 had 5% donor engraftment in bone marrow and P-ML005 had 100% donor engraftment in bone marrow.
ML NK cells are primed consistently and are well tolerated
ML NK cells were successfully isolated and primed from all donor types (supplemental Figure 1A). All patients received the target ML NK cell dose within 30 days of chemotherapy (Table 2; supplemental Figure 1B-C). Patients experienced the expected toxicities of chemotherapy, including cytopenias, febrile neutropenia, and nausea/vomiting (Table 2). During infusion of ML NK cells, 2 patients experienced low-grade fever, which has been observed with ML NK cell administration (Table 2).46 Nonhematologic grade 3 or 4 toxicities were minimal but expected in these heavily treated patients with AML (Table 2). GVHD of the skin (grade 1) was observed in 1 patient (P-ML002), who received a haploidentical HCT and had mild skin GVHD at enrollment. Skin GVHD persisted after ML NK cell infusion and the patient also experienced elevation of liver enzymes concerning for possible liver GVHD. Both skin and liver GVHD resolved with low-dose steroids (<10 mg hydrocortisone equivalent daily) and a single dose of tocilizumab. This patient also received an infusion of CD34-selected donor stem cells at 9 months after ML NK cell infusion for persistent cytopenias and low bone marrow cellularity without evidence of recurrent AML or changes in chimerism, which was expected in the haplo-HCT setting.52,53 After the CD34+ stem cell boost, the patient recovered normal hematopoietic function but developed chronic skin GVHD, unrelated to the prior ML NK cell infusion. No other patients developed GVHD or required additional infusion of stem cells.
Adverse events
| Grade 3-4 adverse events | # (%) patients |
|---|---|
| Hematologic | |
| Anemia | 1 (11) |
| Lymphopenia | 1 (11) |
| Thrombocytopenia | 1 (11) |
| Hemolysis | 1 (11) |
| Other | |
| Fever | 2 (22) |
| Vascular (hypertension) | 2 (22) |
| Gastrointestinal (elevated GGT, ALT or AST) | 3 (33) |
| Grade 3-4 adverse events | # (%) patients |
|---|---|
| Hematologic | |
| Anemia | 1 (11) |
| Lymphopenia | 1 (11) |
| Thrombocytopenia | 1 (11) |
| Hemolysis | 1 (11) |
| Other | |
| Fever | 2 (22) |
| Vascular (hypertension) | 2 (22) |
| Gastrointestinal (elevated GGT, ALT or AST) | 3 (33) |
All grade 3 and 4 adverse events based on CTCAE v4.0 that were possibly, probably, or definitely related to study therapy are listed, along with the number (and percentage) of patients who experienced each toxicity. All patients enrolled were assessed for adverse events.
Combined chemotherapy, DLIs, and donor ML NK cells induced remission in pediatric patients with relapsed AML
Eight evaluable patients were assessed for disease response based on bone marrow biopsy (Table 3). At day 28 after ML NK cells, 6 of 8 patients (75%) had a response with 4 patients (50%) achieving CR and 2 patients (25%) having partial response. At 100 days after infusion, 2 patients continued in CR. One patient (P-ML005) had progression of extramedullary disease, although the bone marrow remained leukemia free. Three patients had progression of disease (PD) between days 28 and 100. At day 100, overall responses (relative to disease status at enrollment) included 2 patients in CR (25%), 1 patient with partial response (12.5%), 4 patients with PD, and 1 patient deceased as a result of PD. One of the patients with PD achieved remission after salvage chemotherapy and proceeded to second HCT. Of the 2 patients in CR at day 100, 1 patient relapsed at day 180 and subsequently achieved remission with salvage chemotherapy followed by a second HCT. One patient remained disease-free >2 years after ML NK cell infusion without any subsequent therapies. At 2 years’ follow-up, overall survival is 42% with 1 patient in continued remission after only ML NK cell therapy (supplemental Figure 2).
Treatment outcomes
| P-ML ID | NK cell dose* (total cells) | Time from FLAG to NK cell infusion | Day 28 disease assessment (% BM blasts) | Day 28 disease response | Day 100 disease assessment (% BM blasts) | Day 100 disease response | Current status |
|---|---|---|---|---|---|---|---|
| 2 | 1.86 × 108 | 26 d | MRD negative | CR | MRD negative | CRi | CD34 boost given, remained in remission >2 y after therapy |
| 4 | 2.19 × 108 | 29 d | Peripheral blasts, BM not assessed* | PD† | NA | NA | Disease progression before day 7 and expired before day 28 |
| 5 | 8.69 × 108 | 25 d | MRD negative | CR | NA | NA | Extramedullary disease progression, off study after day 60, expired |
| 7 | 1.38 × 108 | 18 d | MRD negative | CR | 3 | PR | Proceeded to second transplant, remained alive and disease free |
| 8 | 1.53 × 108 | 16 d | MRD negative | CR | MRD negative | CR | Disease progression at day 180, proceeded to second transplant, remained alive and disease free |
| 9 | 4.26 × 108 | 16 d | 56% | PD | NA | NA | Off study after day 70, expired due to disease progression |
| 10 | 1.17 × 108 | 16 d | 16% | PR | 30 | PD | Expired due to disease progression |
| 11 | 1.4 × 108 | 17 d | 10% | PR | 60 | PD | Expired due to disease progression |
| Summary | 2.81 × 108 (average) | 22 d (average) | 4 MRD negative | 4 CR, 2 PR (75% patients with response) | 2 MRD negative | 2 CR/CRi (25% patients remained in CR) | One patient remained in remission >2 y; 3 alive at 2 y (27.5%) |
| P-ML ID | NK cell dose* (total cells) | Time from FLAG to NK cell infusion | Day 28 disease assessment (% BM blasts) | Day 28 disease response | Day 100 disease assessment (% BM blasts) | Day 100 disease response | Current status |
|---|---|---|---|---|---|---|---|
| 2 | 1.86 × 108 | 26 d | MRD negative | CR | MRD negative | CRi | CD34 boost given, remained in remission >2 y after therapy |
| 4 | 2.19 × 108 | 29 d | Peripheral blasts, BM not assessed* | PD† | NA | NA | Disease progression before day 7 and expired before day 28 |
| 5 | 8.69 × 108 | 25 d | MRD negative | CR | NA | NA | Extramedullary disease progression, off study after day 60, expired |
| 7 | 1.38 × 108 | 18 d | MRD negative | CR | 3 | PR | Proceeded to second transplant, remained alive and disease free |
| 8 | 1.53 × 108 | 16 d | MRD negative | CR | MRD negative | CR | Disease progression at day 180, proceeded to second transplant, remained alive and disease free |
| 9 | 4.26 × 108 | 16 d | 56% | PD | NA | NA | Off study after day 70, expired due to disease progression |
| 10 | 1.17 × 108 | 16 d | 16% | PR | 30 | PD | Expired due to disease progression |
| 11 | 1.4 × 108 | 17 d | 10% | PR | 60 | PD | Expired due to disease progression |
| Summary | 2.81 × 108 (average) | 22 d (average) | 4 MRD negative | 4 CR, 2 PR (75% patients with response) | 2 MRD negative | 2 CR/CRi (25% patients remained in CR) | One patient remained in remission >2 y; 3 alive at 2 y (27.5%) |
Treatment outcomes based on IWG criteria for evaluable patients enrolled in the trial who received immunotherapy. Patient P-ML003 had a phenotypic change from AML to T-cell lymphoblastic leukemia at the start of study therapy and was not included in the efficacy assessment.
MRD, minimal residual disease; NA, not available; PD, progressive disease; PR, partial response.
Patients received between 4 × 106 and 10 × 106 NK cells per kilogram (average, 11 × 106 NK cells per kg). Total dose was calculated as (10 × 106 NK cells per kg) × (weight in kg).
Day 28 PD assigned based on patient’s early disease progression. CRi, complete remission with incomplete count recovery.
Adoptively transferred ML NK cells expand and persist in an immune-compatible environment without exogenous cytokines
To define the long-term potential of ML NK cell expansion and persistence in an immune-compatible environment post-HCT, we used mass cytometry to enumerate total NK cells in patients over time. Peripheral blood and bone marrow were serially assessed after NK cell infusion in each patient. FlowSOM identified 4 lymphocyte metaclusters: CD8 T cells, CD4 T cells, B cells, and NK cells (Figure 2A-B).54 Regulatory T cells (Treg) were gated from the CD4 T-cell metacluster as CD25hiCD127loFoxP3+ (Figure 2A-B). NK cells represented most of the lymphocytes at all time points after infusion in both peripheral blood (Figure 2C-E) and bone marrow (Figure 2F). Percentage of NK cells in the blood remained relatively constant after The infusion, with a significant expansion in the number of NK cells through day 100 (Figure 2D-E). Quantitation of NK cells by mass cytometry mirrored results obtained by standard flow cytometry (supplemental Figure 3). Similar to peripheral blood, NK cells were the dominant lymphocyte population in bone marrow through day 28 with a modest reduction at day 100 (Figure 2F).

NK cells expand in patients treated with DLI and ML NK cell infusion. Peripheral blood and bone marrow samples from patients were analyzed by CyTOF. (A) Representative viSNE demonstrating FlowSOM-gated lymphocyte populations (CD34−CD45+CD14−). (B) Heat map of each marker used to annotate FlowSOM-gated lymphocytes in panel A (C) Density map of lymphocytes at the indicated time points from a representative patient’s peripheral blood mononuclear cells. Inset numbers depict the percentage of each population within the indicated cluster. (D) Total number of cells of each lymphocyte population quantitated by CyTOF across the indicated time points in the peripheral blood of patients. (E-F) Percentage of each lymphocyte population quantitated by CyTOF in the peripheral blood (E) and bone marrow (F). CyTOF data were available for patients P-ML002, P-ML003, P-ML005, P-ML007, P-ML008, P-ML009, and P-ML011. (D-F) Data are expressed as the mean ± standard error. CyTOF, cytometry by time of flight; viSNE, visual stochastic neighbor embedding.
NK cells expand in patients treated with DLI and ML NK cell infusion. Peripheral blood and bone marrow samples from patients were analyzed by CyTOF. (A) Representative viSNE demonstrating FlowSOM-gated lymphocyte populations (CD34−CD45+CD14−). (B) Heat map of each marker used to annotate FlowSOM-gated lymphocytes in panel A (C) Density map of lymphocytes at the indicated time points from a representative patient’s peripheral blood mononuclear cells. Inset numbers depict the percentage of each population within the indicated cluster. (D) Total number of cells of each lymphocyte population quantitated by CyTOF across the indicated time points in the peripheral blood of patients. (E-F) Percentage of each lymphocyte population quantitated by CyTOF in the peripheral blood (E) and bone marrow (F). CyTOF data were available for patients P-ML002, P-ML003, P-ML005, P-ML007, P-ML008, P-ML009, and P-ML011. (D-F) Data are expressed as the mean ± standard error. CyTOF, cytometry by time of flight; viSNE, visual stochastic neighbor embedding.
We next investigated the kinetics of ML NK cells relative to other NK cell populations. ML NK cells have distinct characteristics that distinguish them from CD56bright and CD56dim NK cell populations, defined in our prior study.47 For each patient, visual stochastic neighbor embedding (viSNE) plots were generated by unbiased clustering on memory-associated NK cell markers, KIR, and maturation markers (Figure 3A).47 NK cell subsets were annotated using FlowSOM based on expression of established in vivo differentiated ML NK cell markers (CD56hi, NKp46hi, NKp30hi, DNAM-1hi, CD62L+, NKG2A+, Perforin+, Granzyme B+, and CD11blo; see “Methods”).47 A comparison of NK cells in freshly isolated donor peripheral blood to populations in patient peripheral blood at day 14 after infusion demonstrates a distinct phenotypic change consistent with transition from conventional NK cells (donor) to ML NK cells (patient, day 14; Figure 3B-C). Using this mass cytometry–defined profile, we tracked NK cell subsets in patients for months after adoptive transfer (Figure 3D). At the time of infusion, all NK cells expressed CD25 (supplemental Figure 1A), which was reduced after in vivo differentiation, consistent with previous findings (data not shown).47 ML NK cells comprised most of the NK cells in both peripheral blood and bone marrow for the first month after adoptive transfer, then gradually declined with a concomitant increase in the CD56dim NK-cell population (Figure 3D). ML NK cells exhibited increased expression of CD56, thus activating the receptors CD62L, NKG2A, and Ki-67 (Figure 3E-G), consistent with previous reports.46,47 Notably, ML NK cells maintained a memory-like phenotype at day 28 after infusion with increased CD56, NKG2A, and CD62L compared with CD56dim NK cells (supplemental Figure 4). We examined patient serum for IL-2 and did not observe a systemic increase after DLI (supplemental Figure 5). These results demonstrate that ML NK cells persist for ≥3 months in an immune-compatible environment.
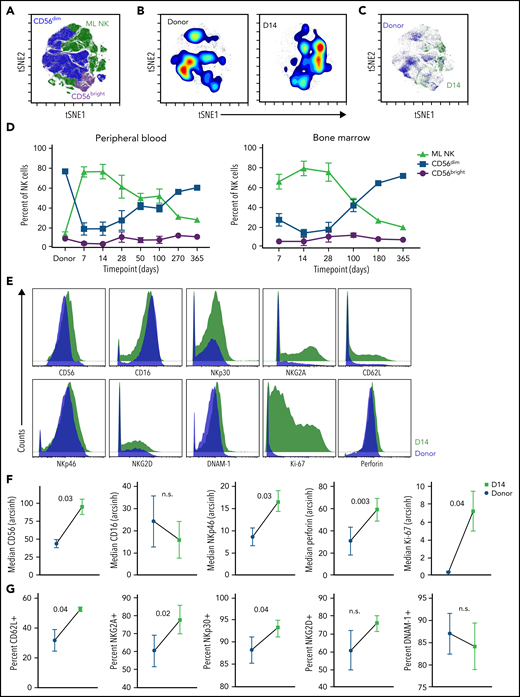
ML NK cells demonstrate prolonged persistence in vivo in patients with relapsed AML. The NK cell multidimensional phenotype was assessed by CyTOF. NK cell subsets were identified by FlowSOM and annotated based on the expression of known ML NK cell markers. (A) Representative composite viSNE demonstrating FlowSOM-gated CD56dim, CD56bright, and ML NK cells from donors and patient peripheral blood at 7 to 60 days after NK cell administration. (B) Density viSNE plot of donor NK cells (at screening) and NK cells from patient peripheral blood at day 14 after NK cell administration. (C) Overlay viSNE from panel B. (D) Summary of NK cell populations across time points in patient peripheral blood and bone marrow. (E) Representative histograms of NK cell markers from donor and patient peripheral blood NK cells at day 14 after infusion. (F-G) Summary of median (F) and percentage positive (G) of indicated markers on donor vs patient peripheral blood NK cells at day 14 after infusion. Data include peripheral blood for P-ML002, P-ML005, P-ML009, and P-ML011, along with bone marrow from P-ML008 (as peripheral blood was not available for this donor). For perforin, P-ML011 was excluded due to a technical failure. (D,F-G) Data are presented as the mean and standard error of the mean. Data were tested for normal distribution (Shapiro-Wilk) and then compared using paired t test or Wilcoxon matched-pairs signed rank test. P-values are indicated. CyTOF, cytometry by time of flight; n.s., not significant; viSNE, visual stochastic neighbor embedding.
ML NK cells demonstrate prolonged persistence in vivo in patients with relapsed AML. The NK cell multidimensional phenotype was assessed by CyTOF. NK cell subsets were identified by FlowSOM and annotated based on the expression of known ML NK cell markers. (A) Representative composite viSNE demonstrating FlowSOM-gated CD56dim, CD56bright, and ML NK cells from donors and patient peripheral blood at 7 to 60 days after NK cell administration. (B) Density viSNE plot of donor NK cells (at screening) and NK cells from patient peripheral blood at day 14 after NK cell administration. (C) Overlay viSNE from panel B. (D) Summary of NK cell populations across time points in patient peripheral blood and bone marrow. (E) Representative histograms of NK cell markers from donor and patient peripheral blood NK cells at day 14 after infusion. (F-G) Summary of median (F) and percentage positive (G) of indicated markers on donor vs patient peripheral blood NK cells at day 14 after infusion. Data include peripheral blood for P-ML002, P-ML005, P-ML009, and P-ML011, along with bone marrow from P-ML008 (as peripheral blood was not available for this donor). For perforin, P-ML011 was excluded due to a technical failure. (D,F-G) Data are presented as the mean and standard error of the mean. Data were tested for normal distribution (Shapiro-Wilk) and then compared using paired t test or Wilcoxon matched-pairs signed rank test. P-values are indicated. CyTOF, cytometry by time of flight; n.s., not significant; viSNE, visual stochastic neighbor embedding.
ML NK cells expand and retain a distinct transcriptional signature
To further define the persistence of ML NK cells, we performed single-cell RNA-sequencing on baseline donor blood NK cells and on the blood samples at several time points after donor NK cell infusion. Notably, NK cells after infusion had a distinct profile compared with NK cells in the donor sample (Figure 4A-B; supplemental Figure 6A-B). Unsupervised Louvain clustering revealed an NK cell population with high concomitant expression of SELL (CD62L), KLRC1 (NKG2A), and FCGR3A (CD16) (Figure 4A-C; supplemental Figure 6A-C), consistent with the ML phenotype identified by mass cytometry (Figure 3; supplemental Figure 4). This ML NK cell cluster is distinct from CD56bright (FCGR3Aneg/lo, TCF7hi), CD56dim (FCGR3Apos), and adaptive (KLRC2hi, KLRC1lo) NK cell clusters (Figure 4A-C; supplemental Figure 6A-C). ML NK cells consist of both KIR+ and KIR− populations and express CD16, further supporting that ML NK cells are not the CD56bright NK cell subset (Figure 4C; supplemental Figure 6C). Consistent with mass cytometry results, ML NK cells expanded comprising ∼50% of NK cells at days 14 to 28 and 23% to 39% of NK cells at 2 months after infusion (Figure 4D; supplemental Figure 6D). These results support that ML NK cells expand, persist, and retain a distinct transcriptional signature after adoptive transfer into an immune-compatible environment.
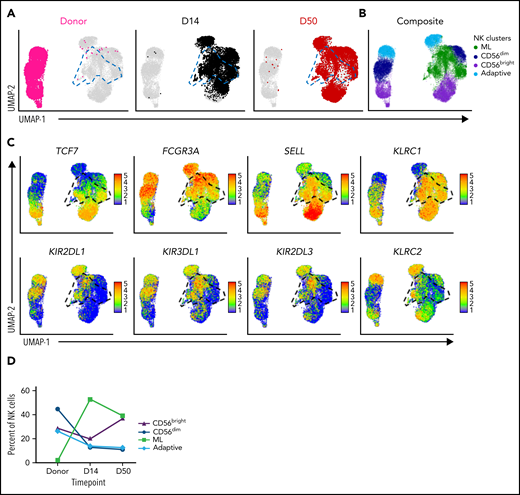
ML NK cells express a unique transcriptional signature in vivo. Single-cell RNA sequencing was performed on enriched NK cells at baseline (donor) and at time points after infusion of cells from patient samples. NK cell subpopulations were identified by unsupervised cluster analysis. Data are shown for peripheral blood for P-ML008. (A) UMAP visualization of NK cells at the indicated time points. Panels are colored by time point overlaid on composite data from all time points (gray). ML NK cells are outlined with blue dashed lines on the UMAPs. (B) UMAP of indicated NK cell populations within composite data from all time points in panel A. (C) Expression of key NK cell population identifying genes. Cells shown in order on the UMAP by expression level. Black gate denotes ML NK cell population. (D) Percentage of CD56bright, CD56dim, ML, and adaptive NK cells in the donor product (baseline, before infusion) and days 14 and 50 after infusion. UMAP, Uniform Manifold Approximation and Projection.
ML NK cells express a unique transcriptional signature in vivo. Single-cell RNA sequencing was performed on enriched NK cells at baseline (donor) and at time points after infusion of cells from patient samples. NK cell subpopulations were identified by unsupervised cluster analysis. Data are shown for peripheral blood for P-ML008. (A) UMAP visualization of NK cells at the indicated time points. Panels are colored by time point overlaid on composite data from all time points (gray). ML NK cells are outlined with blue dashed lines on the UMAPs. (B) UMAP of indicated NK cell populations within composite data from all time points in panel A. (C) Expression of key NK cell population identifying genes. Cells shown in order on the UMAP by expression level. Black gate denotes ML NK cell population. (D) Percentage of CD56bright, CD56dim, ML, and adaptive NK cells in the donor product (baseline, before infusion) and days 14 and 50 after infusion. UMAP, Uniform Manifold Approximation and Projection.
ML NK cells retain enhanced function for at least 4 weeks after adoptive transfer
The antileukemic function of NK cells was measured in the blood and bone marrow by IFN-γ production and CD107a degranulation after stimulation with leukemia target cells ex vivo (Figure 5). These assays revealed that NK cells from patient peripheral blood at multiple time points after adoptive therapy had robust activity, with increased IFN-γ production and CD107a degranulation in response to leukemia (Figure 5B). In addition, antitumor responses were maintained and increased over time, even after proliferation and expansion of the ML NK cells (Figure 5B). Within the same patient sample, ML NK cells demonstrated increased degranulation in response to tumor targets compared with conventional CD56dim NK cells in both peripheral blood and bone marrow (Figure 5C,F). These results demonstrate that the function of ML NK cells is preserved in vivo for weeks in a compatible immune environment.

ML NK cells are highly functional ex vivo. NK cells from patient peripheral blood (A-B) and bone marrow (C-D) were unstimulated or stimulated with K562 in a standard 6-hour functional assay. (A) Representative data depicting IFN-γ and CD107a in unstimulated and K562-stimulated NK cells from P-ML007 peripheral blood at day 28. Numbers represent percentage of cells in the indicated gate. (B) Summary data from patient peripheral blood NK cells stimulated as in panel A indicated over time. Healthy donor NK cells (collected at screening) stimulated with K562 are included as representative of naive NK cell response. (C) Summary data for CD107a degranulation gated on CD56dim (CD56dim NKG2A+/−) or ML (CD56hi NKG2A+) NK cells in the same patient’s peripheral blood sample stimulated as in panel A. (B-C) Data are expressed as the mean ± standard error of the mean. (D) Representative data depicting IFN-γ and CD107a in unstimulated and K562-stimulated NK cells from bone marrow of P-ML007 at day 14. Numbers represent the percentage of cells in the indicated gate. (E) Summary data from bone marrow NK cells of each patient shown at day 14 stimulated as in panel D. (F) Summary data for CD107a degranulation gated on CD56dim (CD56dim NKG2A+/−) or ML (CD56hi NKG2A+) NK cells in same patient bone marrow sample stimulated as in (D). (B-C,E-F) Data were available for P-ML002, P-ML003, P-ML005, and P-ML007. Unstimulated and stimulated conditions were tested for normal distribution (Shapiro-Wilk) then compared by using 2-way analysis of variance (B-C) or paired t test (E-F). P-values are indicated above the graphs.
ML NK cells are highly functional ex vivo. NK cells from patient peripheral blood (A-B) and bone marrow (C-D) were unstimulated or stimulated with K562 in a standard 6-hour functional assay. (A) Representative data depicting IFN-γ and CD107a in unstimulated and K562-stimulated NK cells from P-ML007 peripheral blood at day 28. Numbers represent percentage of cells in the indicated gate. (B) Summary data from patient peripheral blood NK cells stimulated as in panel A indicated over time. Healthy donor NK cells (collected at screening) stimulated with K562 are included as representative of naive NK cell response. (C) Summary data for CD107a degranulation gated on CD56dim (CD56dim NKG2A+/−) or ML (CD56hi NKG2A+) NK cells in the same patient’s peripheral blood sample stimulated as in panel A. (B-C) Data are expressed as the mean ± standard error of the mean. (D) Representative data depicting IFN-γ and CD107a in unstimulated and K562-stimulated NK cells from bone marrow of P-ML007 at day 14. Numbers represent the percentage of cells in the indicated gate. (E) Summary data from bone marrow NK cells of each patient shown at day 14 stimulated as in panel D. (F) Summary data for CD107a degranulation gated on CD56dim (CD56dim NKG2A+/−) or ML (CD56hi NKG2A+) NK cells in same patient bone marrow sample stimulated as in (D). (B-C,E-F) Data were available for P-ML002, P-ML003, P-ML005, and P-ML007. Unstimulated and stimulated conditions were tested for normal distribution (Shapiro-Wilk) then compared by using 2-way analysis of variance (B-C) or paired t test (E-F). P-values are indicated above the graphs.
Discussion
Based on our prior clinical experience with ML NK cells in adults with relapsed/refractory AML, we investigated the feasibility, safety, and preliminary efficacy of donor ML NK cellular immunotherapy to treat AML that relapsed after HCT in pediatric/YA patients. In this approach, ML NK cells are generated from the original HCT donor and thus are adoptively transferred into an immune-compatible environment, yet remain allogeneic to the patient. This provided a unique opportunity to monitor expansion, persistence, and antileukemic activity of these cells over time. Our study sample was small, but the serial assessments of each patient across an extended time course and implementation of mass cytometry and single-cell RNA sequencing assays provide new insight into human ML NK cell biology from the perspective of both the individual and cohorts. Remarkably, we found that after a single dose, ML NK cells persisted in blood and bone marrow for several months (up to 6 months) after transfer and maintained antileukemia activity and ML characteristics. In addition, adoptively transferred donor-derived ML NK cells were well tolerated and, in combination with chemotherapy and DLI-induced clinical responses in highly refractory, heavily pretreated patients with relapsed AML after HCT, 75% of patients responded, with 50% in CR at day 28, and several patients achieving long, durable remissions. Our results provide a platform for future trials in larger cohorts and for future in vivo studies of NK cell immunology.
Adoptive NK cell immunotherapy has been investigated as a potential antileukemia therapy related to established NK cell activity against AML.23-28 Initial trials were based on NK recognition of HLA-mismatching on AML cells in the post-HCT context.29,30,35,36,38 Subsequent studies investigated NK cell function in preventing relapse after HCT based on KIR mismatching between donor and recipient.31-33 Adoptive NK cell therapies have also been investigated using IL-2 or -15 to activate NK cells and boost functional capacity before transfer into patients.35,37,38,55 However, these previous approaches had limited remissions, possibly because of the short window of opportunity for these allogeneic, activated NK cells.24 In the context of AML relapse after HCT, infusion of donor-derived, IL-21–stimulated NK cells or haploidentical NK cells have both been shown to be safe; but, data on efficacy and NK cell persistence and activity after infusion are limited.56,57 We previously established that differentiation of conventional NK cells to ML NK cells by activating their IL-12, -15, and -18 receptors led to more robust antileukemic activity that persisted after adoptive transfer.46,47 Haploidentical ML NK cells in combination with low-dose IL-2 administration induced remission in 47% of adult patients with relapsed/refractory AML in a nontransplant setting. However, as expected, the therapeutic window for ML NK cell activity was limited to 3 weeks, reflecting recovery of the patients’ T cells that rejected donor ML NK cells. Thus, in this clinical setting it was not possible to investigate the persistence and long-term properties of ML NK cells.
In the present study, ML NK cell immunotherapy was used in a post-HCT permissive immune environment, using cells derived from the original transplant donor. Within this context, we established several new characteristics of ML NK cells. First, multidimensional phenotyping revealed ML NK cells expand and persist in patients for ≥3 months; second, ML NK cells retain potent antileukemic responses throughout this extended time; and third, ML NK cells persist without exogenous cytokine support. Collectively, these findings demonstrate that ML NK cells within a normal, compatible immune environment can proliferate and expand while preserving the ML phenotype. Thus, ML NK cells most likely undergo a differentiation program that is maintained through cell division and passed on to daughter cells. These findings are notable when contrasted to the relatively short NK cell half-life and establish a new paradigm for long-lived ML NK cells within human NK cell biology.58 Expansion and persistence of the ML NK cells after infusion may be supported locally by IL-2 from T cells provided by DLI, by other endogenous cytokines (eg, IL-15), or by cell-intrinsic, cytokine-independent signals.48,49 Further investigation of these mechanisms is warranted in future correlative studies as this therapeutic approach progresses. Collectively, these data suggest that strategies to minimize recipient/host rejection of allogeneic ML NK cells may improve expansion, persistence, and clinical outcomes in the allogeneic adoptive therapy setting.
The use of donor-derived ML NK cells in combination with a T-cell-based DLI in the treatment of pediatric and YA patients with relapsed AML after HCT was feasible and safe. ML NK cells were successfully generated from multiple donor sources, including siblings, haploidentical donors, and matched unrelated donors. Notably, sufficient numbers of ML NK cells could be generated from young and older (adult) donors. The feasibility of producing ML NK cells from diverse donor types and ages provides a greater opportunity to extend this immunotherapy to a broader cohort of patients. Furthermore, ML NK cell immunotherapy was well tolerated and safe with little toxicity. Importantly, no patients experienced significant toxicities attributable to the cellular therapy product, such as cytokine release syndrome or immune effector cell-associated neurotoxicity syndrome characteristic of chimeric antigen receptor T cells. Moreover, addition of ML NK cells to standard-of-care chemotherapy and DLI was not associated with an increased incidence of GVHD. Only 1 patient developed mild GVHD, which resolved with minimal therapy. GVHD is an established risk of DLIs, and the low incidence in our cohort suggests that ML NK cells do not substantially increase that risk. Further, ML NK cells may reduce incidence of GVHD as IL-12, -15, and -18–activated NK cells have been shown to reduce acute GVHD in 1 murine model.59 Overall, adoptive transfer of donor-derived ML NK cells was well tolerated and not associated with known toxicities of T-cell-based immunotherapies.
Pediatric/YA AML relapse after HCT remains a significant clinical challenge. In the phase 1 trial reported herein, treatment of patients with relapsed AML with donor ML NK cell therapy in combination with chemotherapy and DLI had an overall response rate of 75% and CR rate of 50% at 28 days after NK cell infusion. One patient has remained in CR without any additional therapies for greater than 2 years and 2 patients with recurrence of disease remain in CR after salvage chemotherapy and repeat HCT. Notably, the enrolled patients disproportionately were heavily pretreated and had AML with very high-risk characteristics. Given this context, the response rates to this novel immunotherapy are remarkably favorable, compared with outcomes with DLI or chemotherapy-alone approaches, in which DLI is successful in <11% of patients.9-17 Chemotherapy approaches are typically used as a bridge to repeat HCT but have limited success in inducing remission and are associated with higher toxicities in high-risk populations, similar to our enrolled patients. Commonly used salvage regimens aimed at achieving a CR in relapsed patients include FLAG (fludarabine, cytarabine, and granulocyte colony-stimulating factor), with or without idarubicin or liposomal daunorubicin, MEC (mitoxantrone, etoposide, and cytarabine), and clofarabine.60-66 In previous studies using these regimens, pediatric patients required at least 2 cycles of chemotherapy to achieve remission.60-64 Overall, these salvage regimens (with 2 cycles of therapy) achieve CR rates of between 40% and 70%, where the higher response rates were in regimens that included anthracyclines. However, a high percentage of patient who achieved CR with these approaches had significant comorbidities, including infection and cardiotoxicity, that precluded proceeding to HCT or increased post-HCT complications.60-64 Newer approaches, using a liposomal preparation of daunorubicin and cytarabine (CPX-351) or combination of venetoclax and cytarabine (with or without idarubicin), have achieved clinical responses after a single cycle of therapy with overall similar outcomes to historic regimens.67,68 Notably, patients who require more than 1 salvage therapy (similar to our enrolled cohort) had even poorer prognosis, with <25% attaining CR.9 Finally, despite achieving CR, overall survival of pediatric patients with relapsed AML, including those that proceed to HCT, is <40%, with relapsed disease being the primary cause of mortality.9,60-64 In our study, combination therapy with chemotherapy, DLI and ML NK cells achieved response rates equivalent to prior chemotherapy only regimens but, in contrast, was associated with minimal toxicities.
Our findings support that ML NK cell immunotherapy is well tolerated, has the potential for inducing remissions in refractory AML relapsed after HCT, and thus warrants further investigation as a cellular therapy for relapsed AML in pediatric/YA patients. Remarkably, in this immune-compatible setting, donor-derived ML NK cells persist for months and show long-term functional activity without the requirement for exogenous cytokines. The use of alternative chemotherapy regimens that may augment ML NK cell activity, or the inclusion of additional infusions to extend ML NK cell persistence may improve the efficacy of this approach.69-72 Assessment of results for patients enrolled with these trial modifications will further advance ML NK cell immunotherapy as a therapeutic option for relapsed AML.
Acknowledgments
Schemas were created by Biorender.com.
This work was supported by National Institutes of Health, National Cancer Institute grants P50CA171963 (T.A.F., M.M.B.-E., and A.F.C.), K12CA167540 (M.M.B.-E.), U01CA248235 (O.L.G. and M.G.), and R01CA205239 (T.A.F.); National Institute of General Medical Sciences Grant T32GM139799 (J.A.F.); National Heart Lung and Blood Institute Grant T32HL00708843 (P.W.); The Children’s Discovery Institute (T.A.F. and J.J.B.); and the American Association of Immunologists: Intersect Fellowship Program for Computational Scientists and Immunologists (J.A.F., A.A.P., and T.A.F.). J.J.B. was supported by an American Society of Hematology Scholar Award, a Gabrielle’s Angel Foundation Award for Cancer Research, and the St. Louis Children’s Hospital Foundation. M.G. was supported by a V Scholar Award (V2018-007) from the V Foundation for Cancer Research; Alvin J. Siteman Cancer Center (NCI grant P30 CA091842) at Washington University School of Medicine and Barnes-Jewish Hospital, Siteman Flow Cytometry Core, Immunomonitoring Laboratory (also supported by the Bursky Center for Human Immunology and Immunotherapy Programs), and the Genome Technology Access Center at McDonnell Genome Institute (also supported by ICTS/CTSA Grant UL1TR002345 from the National Center for Research Resources).
Authorship
Contribution: J.J.B., M.M.B.-E., A.F.C., J.A.F., A.A.P., and T.A.F. designed the research; M.B.-H., S.D., T.S., E.M., M.F., P.P.P., C.C.N., M.M.B.-E., J.A.F., P.W., and S.K.-S. performed the research; J.J.B., C.Z., S.T.B., A.F.C., and T.A.F. enrolled the patients and managed their care during the protocol therapy; J.E. and S.H. collected the clinical data; J.J.B., M.M.B.E., J.A.F., A.A.P., O.L.G., M.G., B.F., W.-R.L., and D.A.R.-G. analyzed the data; T.A.F. supervised the project; J.J.B., T.A.F., M.M.B.E., and J.A.F. wrote the paper; and all authors edited and approved the final manuscript.
Conflict-of-interest disclosure: J.J.B. discloses an advisory role for Horizon Therapeutics. T.A.F. reports grants from HCW Biologics Inc. and the National Institutes of Health (NIH) during the conduct of the study; equity, grants, and consulting fees from Wugen, Inc.; research funding from H.C.W. Biologics, ImmunityBio, Compass Therapeutics, and Affimed; consulting for Kiadis, Nkarta, and Nektar; other support from Indapta and OrcaBio outside the submitted work. T.A.F. and M.M.B.-E. disclose patents (15/983 275, PCT/US 2019/060005, and 62/963 971) pending and licensed to Wugen, Inc.; and equity interest in, consulting for, and royalty interest in Wugen, Inc. This includes intellectual property with T.A.F. and M.M.B.-E. as coinventors, licensed to Wugen, Inc. from Washington University, related to this work. J.A.F. has pending patents (WO 2019/152387, US 63/018 108) unrelated to the present work that are licensed to Kiadis, and a monoclonal antibody unrelated to the present work licensed to EMD Millipore. W.-R.L. is employed by MilliporeSigma. The remaining authors declare no competing financial interests.
Correspondence: Todd A. Fehniger, Washington University School of Medicine, 660 South Euclid, Campus Box 8007, St. Louis, MO 63110; e-mail: tfehnige@wustl.edu; and Jeffrey J. Bednarski, Washington University School of Medicine, 660 South Euclid, Campus Box 8208, St. Louis, MO 63110, e-mail: bednarski_j@wustl.edu.
Sequencing data are available to qualified investigators through dbGAP: phs002681. Selected patient data are included within this manuscript. All individual participant data will not be shared at this time, as the trial is ongoing. Complete deidentified data and the study protocol will be available to qualified investigators upon request at trial completion. Requests can be sent to tfehnige@wustl.edu or bednarski_j@wustl.edu.
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal