

Key Points
DNA hypermethylation of an active intronic CRBN enhancer region downregulates CRBN expression and mediates IMiD resistance in advanced MM.

Abstract
Cereblon is the direct binding target of the immunomodulatory drugs (IMiDs) that are commonly used to treat multiple myeloma (MM), the second most frequent hematologic malignancy. Patients respond well to initial treatment with IMiDs, but virtually all patients develop drug resistance over time, and the underlying mechanisms are poorly understood. We identified an as yet undescribed DNA hypermethylation in an active intronic CRBN enhancer. Differential hypermethylation in this region was found to be increased in healthy plasma cells, but was more pronounced in IMiD-refractory MM. Methylation significantly correlated with decreased CRBN expression levels. DNA methyltransferase inhibitor (DNTMi) in vitro experiments induced CRBN enhancer demethylation, and sensitizing effects on lenalidomide treatment were observed in 2 MM cell lines. Thus, we provide first evidence that aberrant CRBN DNA methylation is a novel mechanism of IMiD resistance in MM and may predict IMiD response prior to treatment.
Introduction
Immunomodulatory drugs (IMiDs) are backbone agents of various multiple myeloma (MM) treatment regimens, with cereblon (CRBN) as the central orchestrator of their antitumor action.1,2 CRBN is a substrate receptor of the CRL4CRBN-E3–ubiquitin ligase complex.1,3 Binding of IMiDs leads to the ubiquitination and degradation of the transcription factors Ikaros (IKZF1) and Aiolos (IKZF3) and the subsequent downregulation of c-Myc and IRF4, which causes inhibition of MM growth and apoptosis.4,5 Reduced levels of or complete loss of CRBN expression, at both the messenger RNA and protein levels, have been associated with IMiD resistance.2,3,6-9 Furthermore, in previous work, we identified underlying genomic mutations in the coding sequence of CRBN.10,11
Epigenetic plasticity is an adaptive way for tumor cells to evade environmental factors such as anticancer therapy. But not much is known about epigenetic mechanisms or posttranscriptional factors that may regulate CRBN expression. Notably, acquired resistance to IMiDs in MM can be reversed in vitro by using a combination of a DNA methyltransferase and an EZH2 inhibitor.12 The addition of oral 5 -azacytidine to lenalidomide-dexamethasone induced meaningful clinical remissions in a lenalidomide-refractory MM cohort.13 Previous studies suggested an epigenetic mechanism of IMiD resistance; however, it was shown that silencing of CRBN by promoter hypermethylation is not the underlying cause of CRBN downregulation.12,14
In this study, we investigated an alternative intragenic CRBN regulatory region (GRCH38: Chr.3 3172800-3173801 ENSR00000675670), which was identified as an active enhancer by its H3K4me1 and H3K27ac histone pattern.15 This novel, yet unexplored enhancer is located downstream of the described 5' CRBN promoter region (GRCH38: Chr.3 3178000-3180601 ENSR00000147822). We analyzed primary samples from patients with newly diagnosed MM (NDMM) and IMiD relapsed or refractory MM (rrMM) as well as plasma cells from healthy volunteers, and we assessed the DNA methylation state of the known CRBN promoter and the novel CRBN enhancer.
Study design
We collected primary MM tumor samples from 131 patients at the Hospital Universitario 12 de Octubre (Madrid, Spain), the University Hospital of Würzburg (Würzburg, Germany), and the University Hospital of Ulm (Ulm, Germany). Of these patients, 79 were newly diagnosed16 and 52 had relapsed MM (rMM), including 27 patients who were refractory to IMiDs (rrMM) according to the International Myeloma Working Group criteria (supplemental Table 4, available on the Blood Web site). We analyzed 45 peripheral blood mononuclear cells (PBMCs) and 52 CD138+ plasma cells from bone marrow aspirates (femoral head) from healthy donors; 26 samples were collected at the University Hospital of Würzburg, 18 at the Hospital Universitario 12 de Octubre, and 8 at the Department of Oncology and Hemato-Oncology, University of Milan (Milan, Italy) and used those samples as controls. An independent control group of 32 white blood cell samples from healthy donors was provided by the Institute of Human Genetics, Würzburg University. Sample collection was performed in accordance with the Declaration of Helsinki, and all participants gave written informed consent.
Plasma cells were enriched from bone marrow aspirates using anti-CD138+ immunomagnetic beads, and PBMCs were separated using gradient centrifugation. DNA was isolated and 100 to 200 ng was used for bisulfite conversion. We analyzed the promoter and enhancer DNA methylation status by using deep bisulfite sequencing on the Illumina MiSeq next-generation sequencer as described elsewhere.17 Real-time quantitative polymerase chain reaction (RT-qPCR) to correlate CRBN methylation and gene expression was performed in a subset of 26 MM samples and in 15 PBMCs with available RNA. Functional validation of the novel CRBN enhancer was confirmed through luciferase reporter assay.18,19 MM cell lines KMS-11 and OPM2 were treated with the demethylating drugs 5-aza-2'-deoxycytidine and 5-azacytidine. Demethylation of the CRBN enhancer region was monitored by pyrosequencing. Lenalidomide sensitivity was measured by annexin V-propidium iodide fluorescence-activated cell sorting. Detailed experimental workup settings are described in the supplemental Methods.
Results and discussion
In epigenome-wide screens, B-cell malignancies usually present global DNA hypomethylation and loci-specific hypermethylation within tumor suppressor promoter regions.20,21 In MM, changes in DNA methylation have been demonstrated to have an impact on pathogenesis, prognosis, and disease progression.21-23 Hypomethylating agents synergize with IMiDs and may even revert resistance; however, the underlying mechanisms have not been fully explained.12,13 Other researchers have demonstrated that the CRBN promoter region (ENSR00000147822) is unmethylated in IMiD-resistant disease in vitro and in vivo.12,14 This was confirmed in 92 of our MM samples and 52 CD138+ plasma cells from healthy donors by using in-house targeted deep bisulfite sequencing methodology,17 in which no promoter hypermethylation was detectable in 39 investigated CpGs (mean methylation, 0.22% ± 0.13% standard deviation; Figure 1). Strikingly, we found remarkable variation in DNA methylation of the novel CRBN enhancer region (ENSR00000675670) located 6039 bp downstream of the promoter in our 131-patient cohort (range, 0.00% to 84.43%). It is noteworthy that this area is not represented on the commonly used Illumina Infinium MethylationEPIC array, which may explain why this hypermethylation remained undetected in previous large-scale epigenome-wide studies. DNA methylation was largely absent in PBMCs (mean, 6.46% ± 3.04%; n = 45) and white blood cells from healthy donors (mean, 4.08% ± 1.11%; n = 32), but it was present in CD138+ plasma cells from healthy donors (24.27% ± 14.95%; n = 52) and CD138+ plasma cells from MM patients (mean, 26.21% ± 23.56%; n = 131; Figure 1). Interestingly, CRBN enhancer methylation significantly increased from NDMM patients (mean, 22.45% ± 19.93%; n = 79) to IMiD-refractory patients (rrMM mean, 40.59% ± 27.65%; n = 27; P = .002). Patients with rMM not refractory to IMiDs, unexposed or with unknown response status (rMM mean, 22.54% ± 23.04%; n = 25), showed significantly lower methylation compared with patients who had rrMM (P = .004) (Figure 2A). The mean methylation of all 131 investigated MM patients (26.21%) was chosen to define the threshold of a hypermethylated state. Accordingly, 39.24% (31 of 79) of NDMM and 66.67% (18 of 27) of IMiD rrMM patients were identified to be hypermethylated.
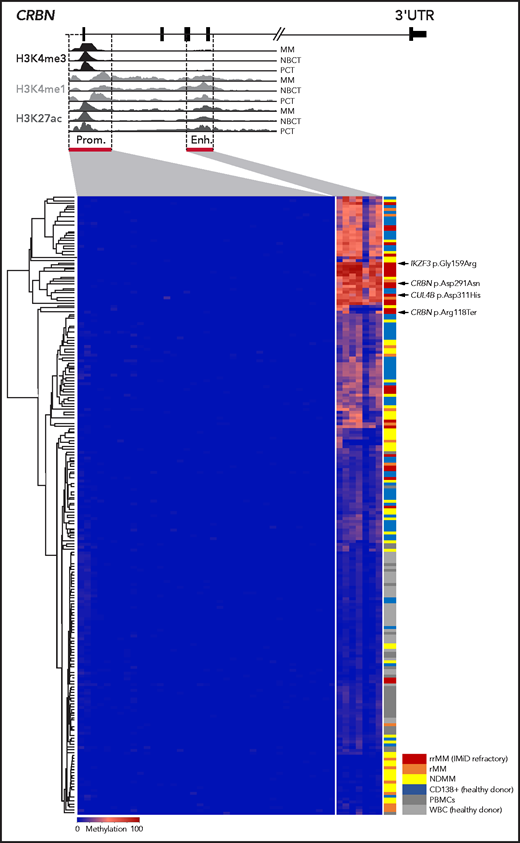
DNA methylation of the CRBN promoter and enhancer in MM patients and CD138+ plasma cells from healthy donors.CRBN has 2 regulatory regions, a 5' promoter (Prom.) and a 6039 bp downstream located active intronic enhancer (Enh.) region as shown in a UCSC plot of representative primary MM as well as naïve B cells from tonsil (NBCT) and plasma cells from tonsil (PCT) from healthy donors. DNA methylation was detected only in the enhancer region and accumulated in IMiD-resistant MM patients. Each line represents an individual sample, and each column represents a single CpG; blue encodes for an unmethylated state, red for a methylated state. The different study cohorts are marked by a second color code. UTR, untranslated region.
DNA methylation of the CRBN promoter and enhancer in MM patients and CD138+ plasma cells from healthy donors.CRBN has 2 regulatory regions, a 5' promoter (Prom.) and a 6039 bp downstream located active intronic enhancer (Enh.) region as shown in a UCSC plot of representative primary MM as well as naïve B cells from tonsil (NBCT) and plasma cells from tonsil (PCT) from healthy donors. DNA methylation was detected only in the enhancer region and accumulated in IMiD-resistant MM patients. Each line represents an individual sample, and each column represents a single CpG; blue encodes for an unmethylated state, red for a methylated state. The different study cohorts are marked by a second color code. UTR, untranslated region.
![CRBN enhancer DNA methylation correlates with gene expression, affects IMiD sensitivity in vitro, and impacts clinical outcome of MM patients exposed to IMiDs. (A) Methylation significantly increased from NDMM to IMiD rrMM (P < .002 ). (B) CRBN enhancer methylation and gene expression showed a significant negative correlation (Spearman’s rho = −0.407; P = .008). For patients with high methylation, gene expression was suppressed. (C) Significantly decreased CRBN enhancer methylation, CRBN expression and lenalidomide (Len) sensitization of KMS-11 (upper panel) and OPM2 cells (lower panel) after exposure to DNMTi’s (500 nM 5-azacytidine [Aza] for 48 hours or 100 nM 5-aza-2′-deoxycytidine [Dec] for 72 hours). Left: CRBN enhancer methylation, measured by pyrosequencing. Center: CRBN expression 5 days after incubation with DNA methyltranferase inhibitors (DNMTi) analyzed by using RT-PCR TaqMan assays, ΔΔCt method, and GUSB as housekeeping control. Right: after pretreatment with DNMTi for 48 hours with 500 nM 5-azacytidine or 72 hours with 100 nM 5-aza-2′-deoxycytidine, cells were treated for 5 days with 10 µM lenalidomide. Cell survival was measured by annexin V-propidium iodide (PI) fluorescence-activated cell sorting. Significantly restored IMiD sensitivity was observed in both cell lines. (D) Functional luciferase activity of CpG-free pCpGL vector containing in vitro methylated or unmethylated CRBN enhancer (Firefly), normalized to internal Renilla control activity in the L363 cell line. Empty MP -pCpGL vector served as a negative control. (E) Kaplan-Meier analysis of NDMM patients who underwent IMiD containing therapies (thalidomide or lenalidomide). Patients with CRBN enhancer (enh.) methylation (meth.) >26% (red) showed significantly inferior PFS compared with patients with methylation levels <26% (blue). Significance level: *P < .05; **P < .01; ***P < .001. DMSO, dimethylsulfoxide; n.s., not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/18/10.1182_blood.2020010452/4/m_bloodbld2020010452f2.png?Expires=1765933725&Signature=3AZfx5VlTH~vMEaP6~TJjIDpjdRWLUyN3YA~XV-5z7NXuHIBkG5MOI7GDIsBuAg9F-OvMk8hryokRKdqHwm60foSY6N8d5XMYbPhuiEF4tUu4htqS6RoLj-t-LdvfBRbJvyOxqLAWXfzI5ai2VnNVHGsnT0LX24rcy34zpkZGfkYbD8svy6DZs7AQF1tuEsfcjApyKHCpri7df7BhHN925MfHNRUbGekmLWDoGJfFoo~1OVfsfUz~tw91v~eRbPctOVQaVQckVsL-FzGuRGKJKzTIPT6w2EZpcHbI1j74oOai2XhiX85ybRq0HNIM0JmHh3Lo3dK9G9~ULqlSd1VIQ__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CRBN enhancer DNA methylation correlates with gene expression, affects IMiD sensitivity in vitro, and impacts clinical outcome of MM patients exposed to IMiDs. (A) Methylation significantly increased from NDMM to IMiD rrMM (P < .002 ). (B) CRBN enhancer methylation and gene expression showed a significant negative correlation (Spearman’s rho = −0.407; P = .008). For patients with high methylation, gene expression was suppressed. (C) Significantly decreased CRBN enhancer methylation, CRBN expression and lenalidomide (Len) sensitization of KMS-11 (upper panel) and OPM2 cells (lower panel) after exposure to DNMTi’s (500 nM 5-azacytidine [Aza] for 48 hours or 100 nM 5-aza-2′-deoxycytidine [Dec] for 72 hours). Left: CRBN enhancer methylation, measured by pyrosequencing. Center: CRBN expression 5 days after incubation with DNA methyltranferase inhibitors (DNMTi) analyzed by using RT-PCR TaqMan assays, ΔΔCt method, and GUSB as housekeeping control. Right: after pretreatment with DNMTi for 48 hours with 500 nM 5-azacytidine or 72 hours with 100 nM 5-aza-2′-deoxycytidine, cells were treated for 5 days with 10 µM lenalidomide. Cell survival was measured by annexin V-propidium iodide (PI) fluorescence-activated cell sorting. Significantly restored IMiD sensitivity was observed in both cell lines. (D) Functional luciferase activity of CpG-free pCpGL vector containing in vitro methylated or unmethylated CRBN enhancer (Firefly), normalized to internal Renilla control activity in the L363 cell line. Empty MP -pCpGL vector served as a negative control. (E) Kaplan-Meier analysis of NDMM patients who underwent IMiD containing therapies (thalidomide or lenalidomide). Patients with CRBN enhancer (enh.) methylation (meth.) >26% (red) showed significantly inferior PFS compared with patients with methylation levels <26% (blue). Significance level: *P < .05; **P < .01; ***P < .001. DMSO, dimethylsulfoxide; n.s., not significant.
CRBN enhancer DNA methylation correlates with gene expression, affects IMiD sensitivity in vitro, and impacts clinical outcome of MM patients exposed to IMiDs. (A) Methylation significantly increased from NDMM to IMiD rrMM (P < .002 ). (B) CRBN enhancer methylation and gene expression showed a significant negative correlation (Spearman’s rho = −0.407; P = .008). For patients with high methylation, gene expression was suppressed. (C) Significantly decreased CRBN enhancer methylation, CRBN expression and lenalidomide (Len) sensitization of KMS-11 (upper panel) and OPM2 cells (lower panel) after exposure to DNMTi’s (500 nM 5-azacytidine [Aza] for 48 hours or 100 nM 5-aza-2′-deoxycytidine [Dec] for 72 hours). Left: CRBN enhancer methylation, measured by pyrosequencing. Center: CRBN expression 5 days after incubation with DNA methyltranferase inhibitors (DNMTi) analyzed by using RT-PCR TaqMan assays, ΔΔCt method, and GUSB as housekeeping control. Right: after pretreatment with DNMTi for 48 hours with 500 nM 5-azacytidine or 72 hours with 100 nM 5-aza-2′-deoxycytidine, cells were treated for 5 days with 10 µM lenalidomide. Cell survival was measured by annexin V-propidium iodide (PI) fluorescence-activated cell sorting. Significantly restored IMiD sensitivity was observed in both cell lines. (D) Functional luciferase activity of CpG-free pCpGL vector containing in vitro methylated or unmethylated CRBN enhancer (Firefly), normalized to internal Renilla control activity in the L363 cell line. Empty MP -pCpGL vector served as a negative control. (E) Kaplan-Meier analysis of NDMM patients who underwent IMiD containing therapies (thalidomide or lenalidomide). Patients with CRBN enhancer (enh.) methylation (meth.) >26% (red) showed significantly inferior PFS compared with patients with methylation levels <26% (blue). Significance level: *P < .05; **P < .01; ***P < .001. DMSO, dimethylsulfoxide; n.s., not significant.
Next, we addressed the question of whether CRBN enhancer hypermethylation has an impact on expression and thus may mediate drug resistance. In fact, we confirmed in 26 of our MM patients and in 15 PBMCs with available RNA that the CRBN enhancer methylation degree correlated significantly with decreased CRBN gene expression (Spearman’s r = −0.406; P = .008; Figure 2B). To explore IMiD desensitizing effects by using demethylating agents, we selected 2 tMM cell lines with intrinsic CRBN enhancer hypermethylation (OPM2 and KMS-11). After exposure to DNMTi (48 hours with 500 nM 5-azacytidine or 72 hours with 100 nM 5-aza-2′-deoxycytidine), we observed a significant reduction of the CRBN enhancer methylation (P < .001). As we had hypothesized, cells demonstrated significantly increased sensitivity to lenalidomide (P < .01) (Figure 2C), and significantly increased expression of CRBN was assessed by qPCR in OPM2 (OPM2 5-aza-2'-deoxycytidine+, P = .013; OPM2 5-azacytidine+, P < .001). However, in KMS-11 cells, the positive trend did not reach statistical significance. Finally, we validated the regulatory properties of the novel CRBN enhancer at a functional level by using a dual-luciferase reporter assay, in which the unmethylated insert demonstrated a significant 34.5-fold increased luciferase activity compared with the methylated insert (P < .001; Figure 2D). Together these results strongly suggest yet undescribed epigenetic regulation of CRBN impacting IMiD response in MM.
Patients with MM are commonly treated with combinations of multiple drugs, which makes it challenging to retrospectively assess the clinical relevance of CRBN enhancer methylation, in particular the degree of IMiD impairment for individual patients. Nevertheless, among the top 5 methylated patients in our cohort for whom clinical information was available, we found confirming evidence of IMiD-resistant disease: 1 patient was IMiD naïve at the time of sampling and had 71% DNA methylation before second-line therapy with carfilzomib-lenalidomide-dexamethasone to which she progressed unexpectedly early after not more than 4 cycles. One patient was primary IMiD refractory with no response to first-line lenalidomide-doxorubicin-dexamethasone. She had 71% methylation when she had no response to pomalidomide-dexamethasone therapy. A patient with 72% methylation receiving fourth-line therapy did not respond to subsequent pomalidomide-bortezomib-doxorubicin-dexamethasone, and another patient with 82% methylation receiving eleventh-line therapy did not respond to either pomalidomide-bortezomib-doxorubicin-dexamethasone or later dosing with elotuzumab-thalidomide-dexamethasone. Still, one patient with 83% CRBN enhancer methylation was initially responsive to first-line lenalidomide-doxorubicin-dexamethasone induction and third-line bortezomib-doxorubicin-lenalidomide-dexamethasone but progressed under fourth-line lenalidomide-elotuzumab-dexamethasone and had no response to fifth-line pomalidomide and subsequent pomalidomide-bortezomib-doxorubicin-dexamethasone or daratumumab-carfilzomib-lenalidomide-dexamethasone.
In a progression-free survival (PFS) analysis, NDMM patients who underwent IMiD based therapy and that had a hypermethylated CRBN enhancer status faced an inferior PFS compared with those who had no CRBN enhancer hypermethylation (median PFS, CRBN methylation >26% at 17 months; 95% confidence interval [CI], 10-23 months vs CRBN methylation <26% at 74 months; 95% CI, 21-127 months; P = .004) (Figure 2E). Notably, this effect was absent in patients who had not received IMiD first-line therapy (supplemental Figure 2).
Further research is needed to determine the clinical effects of CRBN enhancer regulation on IMiD sensitivity. In addition, the druggable properties of the epigenetic modifications need to be further explored. Importantly, epimutations and genetic mutations that we described earlier to inhibit CRBN-IMiD interactions10 were not mutually exclusive in 4 of our patients with available mutation status from M3P analysis24,25 (supplemental Table 5). This provides evidence that both mechanisms in parallel contribute to IMiD resistance in MM.
Acknowledgments
The authors acknowledge Michael Rehli (University Hospital Regensburg, Regensburg, Germany), who kindly provided the empty pCpGL vector and the pCpGL-CMV/EF1 vector, and Ramya Potabattula (Institute of Human Genetics, Julius Maximilians University Würzburg, Würzburg, Germany) for technical advice.
This study was supported by “Stiftung zur Förderung der Krebsforschung an der Universität Würzburg,” Stifterverband für die Deutsche Wissenschaft, “Mildred Scheel Early Career Center” (MSNZ Würzburg) founded by German Cancer Aid (Deutsche Krebshilfe), Centro de Investigación Biomédica en Red-Cáncer (CIBERONC) (CB16/12/00489) cofinanced with Fondo Europeo de Desarrollo Regional (FEDER) funds, and by grants from the European Research Council under the European Union’s Horizon 2020 Research and Innovation Program (no. 817997).
Authorship
Contribution: S.B., L.H., T.H., K.M.K., and L.R. designed the research; S.B., A.G.-T., L.H., S.H., U.M., and C.V. performed the experiments; X.A. and J.I.M.-S. helped with data analysis and gave technical support; M.B., N.B., M.C., M.D.V., H.E., R.A.F., J.K., A.R., M.K., K.M.K., J.M.-L., L.R., P.R., T.S., Y.R.-H., J.Z., and X.Z. provided patient samples and clinical data; L.H., S.B., L.R., and K.M.K. wrote the manuscript which was approved by all authors; and all authors analyzed and interpreted the data.
Conflict-of-interest disclosure: S.B. and Y.R.-H. are employees of and S.B. and J.M.-L. are equity shareholders of Altum Sequencing Co. The remaining authors declare no competing financial interests.
Correspondence: K. Martin Kortüm, Department of Hematology and Oncology, University Hospital Würzburg, Oberdürrbacherstr. 6, 97080 Würzburg, Germany; e-mail: kortuem_m@ukw.de.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal