

Key Points
Mim8, an anti-FIXa/anti-FX bispecific antibody, has enhanced activity over emicizumab in mouse models and in vitro hemophilia A assays.
The activity of Mim8 is partly achieved through FIXa stimulatory activity residing in the anti-FIXa arm.

Abstract
Hemophilia A is a bleeding disorder resulting from deficient factor VIII (FVIII), which normally functions as a cofactor to activated factor IX (FIXa) that facilitates activation of factor X (FX). To mimic this property in a bispecific antibody format, a screening was conducted to identify functional pairs of anti-FIXa and anti-FX antibodies, followed by optimization of functional and biophysical properties. The resulting bispecific antibody (Mim8) assembled efficiently with FIXa and FX on membranes, and supported activation with an apparent equilibrium dissociation constant of 16 nM. Binding affinity with FIXa and FX in solution was much lower, with equilibrium dissociation constant values for FIXa and FX of 2.3 and 1.5 µM, respectively. In addition, the activity of Mim8 was dependent on stimulatory activity contributed by the anti-FIXa arm, which enhanced the proteolytic activity of FIXa by 4 orders of magnitude. In hemophilia A plasma and whole blood, Mim8 normalized thrombin generation and clot formation, with potencies 13 and 18 times higher than a sequence-identical analogue of emicizumab. A similar potency difference was observed in a tail vein transection model in hemophilia A mice, whereas reduction of bleeding in a severe tail-clip model was observed only for Mim8. Furthermore, the pharmacokinetic parameters of Mim8 were investigated and a half-life of 14 days shown in cynomolgus monkeys. In conclusion, Mim8 is an activated FVIII mimetic with a potent and efficacious hemostatic effect based on preclinical data.
Introduction
Treatment options for people with hemophilia A (HA) have for the past 2 decades improved markedly. Recombinant factor VIII (FVIII) molecules have eliminated the risk of viral transmission, and extended half-life FVIII products have further reduced the treatment burden of prophylaxis.1 However, a serious complication of FVIII treatment of HA is the development of neutralizing antibodies (inhibitors) in ∼30% of patients,2 as well as the risk and inconvenience associated with intravenous (IV) administration. Likewise, until recently, the bypassing agents available for the treatment of HA patients with inhibitors have been intravenously administered products (recombinant activated FVII and activated prothrombin complex concentrate) and with limited options available for prophylactic therapy due to their short half-lives in circulation.3 With the launch of the activated FVIII (FVIIIa)-mimetic bispecific antibody (biAb) emicizumab (Hemlibra [Genentech]), the first subcutaneous (SC) prophylactic treatment became available. Emicizumab mimics the effect of FVIIIa by binding to activated factor IX (FIXa) and factor X (FX),4 and it has exhibited good efficacy for prophylactic treatment of HA patients with or without inhibitors.5,6 Even though both FVIIIa and FVIIIa-mimetics stimulate FIXa-mediated activation of FX, there are differences in their modes of action,7 and the cofactor activity of FVIIIa is considerably greater than that of emicizumab.8 With this in mind, the current study aimed to design a highly potent and efficacious FVIIIa-mimetic antibody.
The biology of FVIII to be recapitulated in an FVIIIa-mimetic biAb format is complex. In circulation, FVIII exists predominantly as a heterodimeric pro-cofactor tightly bound to its carrier protein, von Willebrand factor, which shields it from untimely engagement with the components of the coagulation system.9-11 Upon proteolytic activation at the site of injury, von Willebrand factor is released and the resulting FVIIIa localizes to the activated platelet surface,9 where it combines with FIXa to form the intrinsic tenase complex. Compared with free FIXa, the catalytic efficiency of this complex seems to result from improved membrane localization,12,13 optimal proximity and alignment of FIXa and FX for substrate cleavage to occur,14,15 and the allosteric maturation of the active site of FIXa.16-19 Together, these mechanisms enhance the rate of FX activation by 4 to 6 orders of magnitude.20
As shown with emicizumab, the biAb format is well suited to mimic the ability of FVIIIa to bridge protease and substrate.21 We further hypothesized that the allosteric component of the FVIIIa cofactor activity could be recapitulated in the biAb through directed engineering of the anti-FIXa arm. Guided by these considerations, anti-FIXa/anti-FX biAbs were generated in vitro by using the controlled antigen-binding fragment (Fab)-arm exchange (DuoBody [Genmab]) technology (Figure 1A).22 Combined with extensive mutational optimization, Mim8 was created. Here, we report the development of Mim8 as a next-generation FVIIIa mimetic with highly efficient FIXa-stimulating capability, a circulating half-life of 14 days in the cynomolgus monkey, and low target binding in circulation.
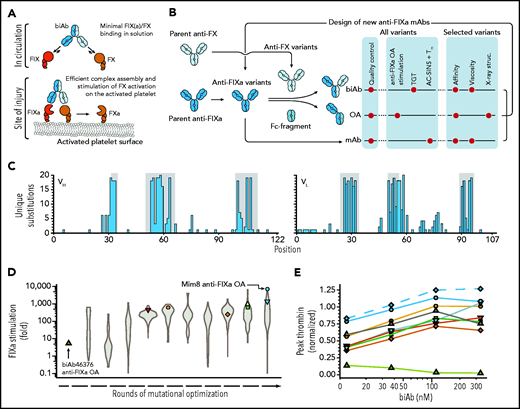
High-throughput screening for FVIIIa-mimetic activity. An overview of the pursued mechanism of action of Mim8 (A) and screening and optimization strategy (B) is shown. Assays used to investigate functional, structural, and biophysical properties of biAb, anti-FIXa OA, and mAb variants are indicated. (C) Number of unique amino acid substitutions explored at individual positions (using consecutive numbering) in the heavy (VH) and light (VL) chain variable domains of the Mim8 anti-FIXa arm. Complementarity-determining region (CDR) loops are highlighted in gray. (D) Evolution of FIXa stimulation of variants of the parental Mim8 anti-FIXa arm during rounds of mutational optimization. FIXa stimulation was measured in high throughput at pH 7.4 in the presence of 0.15 to 1 nM FIXa, 100 nM FX, 500 µM PS:PC vesicles, and a single concentration (800 nM) of anti-FIXa OA. A total of 1308 variants were investigated, with the distribution of activities within each round shown as a violin plot. (E) Plasma activity of a representative subset of biAbs from cycles of anti-FIXa arm optimization. Variant anti-FIXa arms (identified by symbol and color coding as in panel D) were assembled with the parental anti-FX arm. Activity (mean, n = 2) was measured in a TF-triggered thrombin generation assay in human congenital HA plasma at 4 biAb concentrations and normalized to the response obtained with 333 nM emicizumab SIA. Green triangles and blue circles represent biAbs containing the parental (biAb46376) and final Mim8 anti-FIXa arm, respectively. The final engineered Mim8 is included for reference and shown by blue diamonds and a dashed line.
High-throughput screening for FVIIIa-mimetic activity. An overview of the pursued mechanism of action of Mim8 (A) and screening and optimization strategy (B) is shown. Assays used to investigate functional, structural, and biophysical properties of biAb, anti-FIXa OA, and mAb variants are indicated. (C) Number of unique amino acid substitutions explored at individual positions (using consecutive numbering) in the heavy (VH) and light (VL) chain variable domains of the Mim8 anti-FIXa arm. Complementarity-determining region (CDR) loops are highlighted in gray. (D) Evolution of FIXa stimulation of variants of the parental Mim8 anti-FIXa arm during rounds of mutational optimization. FIXa stimulation was measured in high throughput at pH 7.4 in the presence of 0.15 to 1 nM FIXa, 100 nM FX, 500 µM PS:PC vesicles, and a single concentration (800 nM) of anti-FIXa OA. A total of 1308 variants were investigated, with the distribution of activities within each round shown as a violin plot. (E) Plasma activity of a representative subset of biAbs from cycles of anti-FIXa arm optimization. Variant anti-FIXa arms (identified by symbol and color coding as in panel D) were assembled with the parental anti-FX arm. Activity (mean, n = 2) was measured in a TF-triggered thrombin generation assay in human congenital HA plasma at 4 biAb concentrations and normalized to the response obtained with 333 nM emicizumab SIA. Green triangles and blue circles represent biAbs containing the parental (biAb46376) and final Mim8 anti-FIXa arm, respectively. The final engineered Mim8 is included for reference and shown by blue diamonds and a dashed line.
Methods
Human material
Congenital HA plasma (FVIII level <1%) was obtained from George King Bio-Medical Inc. Peripheral blood was drawn from healthy volunteers (approved by “De videnskabsetiske komiteer for Region Hovedstaden”; VEK journal no. H-D-2007-0055) and stabilized by 3.2% (w/v) citrate. Platelet-rich plasma was prepared from citrate-stabilized blood by centrifugation for 10 minutes at 200g.
Variant generation
Anti-FIXa and anti-FX antibodies were produced by using the Expi293 expression system (HEK; Thermo Fisher Scientific). Subsequent assembly of biAbs was performed as described by Labrijn et al23 and detailed in the supplemental Methods (available on the Blood Web site). Generation of anti-FIXa one-arm (OA) antibodies followed the same protocol, except that assembly was performed with a fragment crystallizable (Fc) fragment instead of an anti-FX monoclonal antibody (mAb).
Enzyme kinetic analyses
The activities of biAb and anti-FIXa OA antibodies were measured at room temperature by using plasma-derived (pd) FIXa and FX and synthetic 25:75 PS:PC (25% phosphatidylserine, 75% phosphatidylcholine) vesicles (all from Haematologic Technologies Inc. [HTI]) in reaction buffer (50 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 100 mM NaCl, 5 mM CaCl2, 0.1% PEG8000, and 1 mg/mL bovine serum albumin at pH 7.3). Reactions were quenched by addition of EDTA to a final total concentration of 20 mM. Generated activated FX (FXa) was quantified from the hydrolysis of 500 µM of chromogenic substrate S-2765 (Chromogenix) based on a standard curve with known concentrations of FXa. Details of individual assays and data analysis are provided in the supplemental Methods.
Binding studies and epitope mapping
Direct binding studies were performed by using isothermal titration calorimetry (ITC) at 25°C or 37°C in 10 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid, 150 mM NaCl, and 5 mM CaCl2, pH 7.4. Key residues involved in the recognition of FIXa by the anti-FIXa arm in Mim8 were determined by mutagenesis and subsequent evaluation of binding by surface plasmon resonance analysis. Detailed assay descriptions are provided in the supplemental Methods.
X-ray crystallography
The supplemental Methods provide a detailed description of the crystallization and structure elucidation of the complexes between the anti-FIXa and anti-FX Fab fragments of Mim8 and EGR-chloromethylketone active-site inhibited des-(Gla-EGF1) FIXa and FXa, respectively.
Thrombin generation in human plasma
BiAb activity was measured in a thrombin generation test using pd-FXIa (HTI) or tissue factor (TF) (PRP-Reagent LOW; Thrombinoscope) as trigger and reagents, calibrators, and software from Thrombinoscope. A detailed assay description is provided in the supplemental Methods.
Thromboelastography in human whole blood
Clot formation in human whole blood with antibody-induced HA was determined with TF as trigger (200 000-fold dilution of Innovin) using a Thrombelastograph Coagulation Analyzer (Haemoscope Corporation). Antibodies were tested at final whole blood concentrations of 0.005 to 175 nM (ie, 0.01-350 nM in plasma assuming a hematocrit of 50%). Clot time (R-time) and clot development (α-angle) were calculated by the accompanying software and the concentration-dependent change in R-time fitted to a variable-slope, 4-parameter dose-response model.
Effect studies in HA mouse injury models
HA mice (C57B6/S129 mixed background, 12-16 weeks of age, male and female subjects; The Jackson Laboratory) were anesthetized and placed on a heating pad with tails submerged in tempered saline. To overcome the lack of cross-reactivity of Mim8 with mouse FIXa and FX, HA mice were supplemented with human recombinant FIX (BeneFix, Pfizer Inc.) and pd-FX (HTI) to approximately twice the normal level in humans. Tail vein transection (TVT) studies were performed as previously described24 with a 40-minute duration of bleeding. Tail clip bleeding was performed as noted earlier, except that the entire tail was cut 4 mm above the tail tip, and the duration of bleeding was limited to 30 minutes. Details on the measurement of blood loss and plasma components and data analysis are provided in the supplemental Methods. All studies were approved by the Danish Animal Experimentation Council.
Pharmacokinetic parameters in cynomolgus monkeys
The pharmacokinetic study in the cynomolgus monkey was performed at Covance Ltd. (UK) in accordance with the EU Directive 2010/63/EU for animal experiments and The Animals (Scientific Procedures) Act and Covance Standard Operating Procedures, and it was approved by the Novo Nordisk Ethical Review Committee. Additional details of the study are provided in in the supplemental Methods.
Results
Screening and optimization of anti-FIXa and anti-FX arms
Antibodies against active-site inhibited FIXa and zymogen FX were selected based on immunizations in Kymouse mice (Kymab Ltd.) with a human antibody repertoire25 and from selections using a phage library displaying human antibodies. Subsequent introduction of the K409R (anti-FIXa mAb) or F405L (anti-FX mAb) substitutions in immunoglobulin G1 format allowed for in vitro assembly of biAbs using controlled Fab-arm exchange.23 Due to variability in affinities of the assembled biAbs, functional screening was performed at several concentrations to cover differences in activity optima as a result of the so-called hook effect. This effect is evident as a decline in activity above a certain biAb concentration due to the sequestration of FIXa and FX on separate biAbs and thereby blocking the assembly of productive biAb-FIXa-FX complexes (discussed also by Douglass et al26).
Screening of individual constituents of the 100 × 100 anti-FIXa/anti-FX library for the ability to promote thrombin generation in congenital HA plasma identified several biAbs that increased thrombin levels above baseline. Of these, biAb46376 was selected as the initial lead. It is composed of human anti-FIXa and anti-FX arms from Kymouse and phage display, respectively, and was subsequently reformatted to the immunoglobulin G4 (IgG4) isotype. In the screening assay, the maximum peak thrombin level was observed at 12.3 nM biAb46376, reaching 14% of the level observed for a sequence-identical analogue (SIA) of emicizumab at a clinically relevant concentration of 333 nM6 (Figure 1E; green triangles) (supplemental Methods). Consistent with the early activity optimum for this biAb, ITC experiments showed that the 2 constituent arms bind to FIXa and FX in solution with relatively low equilibrium dissociation constant (KD) values of 1.7 ± 0.5 nM and 101 ± 12 nM, respectively (supplemental Table 2). To investigate the direct stimulation of FIXa by the anti-FIXa arm itself, this arm was prepared in monovalent OA format by assembly with a Fab-less Fc fragment as described earlier for the biAbs.27 This was done to avoid avidity effects with the standard bivalent mAb format resulting in a bell-shaped activity dependence (hook) as also seen for the biAbs. In the OA format, the anti-FIXa arm enhanced the FIXa-mediated activation of FX fourfold (Figure 1D, green triangle).
With these data suggesting that FIXa stimulation contributes to the activity of biAb46376, we sought to further enhance the stimulatory activity of the anti-FIXa arm through mutagenesis. As depicted in Figure 1C, mutagenesis focused primarily on the 6 complementarity-determining region loops, initially addressing a single or a few positions. Substitutions were then combined in a data-driven and iterative fashion to further optimize activity and address any biophysical liabilities. In total, 4056 variants of the anti-FIXa arm were explored individually. The workflow for high-throughput variant generation and screening included determination of biAb and anti-FIXa OA activity in a thrombin generation test and FIXa stimulation assays, respectively. In addition, the thermal melting point and propensity for self-association (using affinity-capture self-interaction nanoparticle spectroscopy [AC-SINS]28) were measured for each anti-FIXa mAb to capture potential developability issues (Figure 1B). The evolution of FIXa stimulation and biAb activity during rounds of engineering are shown in Figure 1D-E. In the high-throughput screening assays, the mutations in the anti-FIXa arm enhanced the stimulation of FIXa >1000 times, relative to the parental biAb46376 anti-FIXa arm (Figure 1D, blue circle), and brought the activity of the biAb in the context of the parental anti-FX arm above that of emicizumab SIA (Figure 1E, blue circles).
The major aim of the optimization of the anti-FX arm was to reduce the affinity to FX to minimize target-mediated clearance and flatten out the bell-shaped activity–concentration dependence generally observed for biAbs with this arm. Following rounds of mutagenesis, the KD value for FX binding was increased from 101 ± 12 nM to 1.5 ± 0.4 µM (supplemental Table 2) while maintaining biAb activity (Figure 1E; blue diamonds). Combining this arm with the optimized anti-FIXa arm using the DuoBody technology in the IgG4 background resulted in the final Mim8 biAb.
Structure determination of Mim8 Fab-FIXa and -FX complexes
The crystal structures of the Mim8 anti-FIXa and anti-FX Fab fragments in complex with EGR- chloromethylketone active-site inhibited des-(gla-EGF1) FIXa or FXa were determined at 3.1 and 2.6 Å resolution, respectively. As shown in Figure 2A,C, the anti-FIXa Fab contacts a small 152-Å2 epitope (4-Å distance cutoff) on FIXa composed of residues from the 170-loop (positions 340-343 using standard FIX numbering) and preceding 160-helix (334-339) as well as a single residue (354) in activation loop 2. Of these, T340 and K341 and in particular R338 on FIXa are important for binding as determined by mutagenesis and surface plasmon resonance analysis (Figure 2C, right panel). Because the anti-FIXa arm binds FIXa on the backside of the 170-loop (ie, away from the active site), it allows for substrate docking in the active site without any apparent steric clashes. Comparison with the crystal structure of the complex between FIXa and antithrombin also shows unimpeded access of antithrombin.30
![Crystal structures of Mim8 anti-FIXa and anti-FX Fab fragments in complex with their respective targets. Crystal structures of Mim8 anti-FIXa (A) and anti-FX Fab (B) fragments in complex with EGR-chloromethylketone active-site inhibited des-(Gla-EGF1) FIXa and FXa, respectively, are shown. Surface representations of the missing Gla and EGF1 domains were included based on the crystal structure of the corresponding domains from full-length human FVIIa29 and with orientations obtained from a structural alignment of the respective EGF2-catalytical domains. (C) Close-up of the anti-FIXa Fab (blue cartoon representation; complementarity-determining region [CDR] loops in gray color). Neighboring residues in FIXa are shown in yellow or red as backbone stick representations with side chains included for residues 338 to 341 and 354. The relative impact of each residue on human FIX binding was evaluated by surface plasmon resonance analysis following amino acid replacement as indicated (right panel) and with the most important residues colored red. Results are shown as residual binding (mean ± standard deviation, n = 9). For all panels, the EGR-chloromethylketone inhibitor (light gray) and calcium ions (light green) are displayed as sphere representations.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/14/10.1182_blood.2020010331/4/m_bloodbld2020010331f2.png?Expires=1762521959&Signature=igz-geO-M--qmpbNDX2teSGbvPsAgQglAezXDJPIIKjdk76pzSMquBuuphRDt6uVjs0Na64jl5wACE8nkIauJdPSXDX1jEa9NsULlSmvAj4urUhzg2vT3yA97FuMzPt3dw2IvjXfPW0OTUgx6bQUclFZoJ0c3GUrcgecxqs2Ihmoh6QAbfkK5erNIdmYHm2-kkO2eYyMq-y9YV5uSo0pszOsPT--ZQOJrm3~ccnG5JaXPWVvHl~z3d99i-qz9auApjFw9xIuul1H7zj8L3a9Vl-pwYaLnrZGGi-M1mYKuAIttKZDnMjnhEvKPvgFy1TzUpHbs9~mWzI~lXsO1z20Sg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Crystal structures of Mim8 anti-FIXa and anti-FX Fab fragments in complex with their respective targets. Crystal structures of Mim8 anti-FIXa (A) and anti-FX Fab (B) fragments in complex with EGR-chloromethylketone active-site inhibited des-(Gla-EGF1) FIXa and FXa, respectively, are shown. Surface representations of the missing Gla and EGF1 domains were included based on the crystal structure of the corresponding domains from full-length human FVIIa29 and with orientations obtained from a structural alignment of the respective EGF2-catalytical domains. (C) Close-up of the anti-FIXa Fab (blue cartoon representation; complementarity-determining region [CDR] loops in gray color). Neighboring residues in FIXa are shown in yellow or red as backbone stick representations with side chains included for residues 338 to 341 and 354. The relative impact of each residue on human FIX binding was evaluated by surface plasmon resonance analysis following amino acid replacement as indicated (right panel) and with the most important residues colored red. Results are shown as residual binding (mean ± standard deviation, n = 9). For all panels, the EGR-chloromethylketone inhibitor (light gray) and calcium ions (light green) are displayed as sphere representations.
Crystal structures of Mim8 anti-FIXa and anti-FX Fab fragments in complex with their respective targets. Crystal structures of Mim8 anti-FIXa (A) and anti-FX Fab (B) fragments in complex with EGR-chloromethylketone active-site inhibited des-(Gla-EGF1) FIXa and FXa, respectively, are shown. Surface representations of the missing Gla and EGF1 domains were included based on the crystal structure of the corresponding domains from full-length human FVIIa29 and with orientations obtained from a structural alignment of the respective EGF2-catalytical domains. (C) Close-up of the anti-FIXa Fab (blue cartoon representation; complementarity-determining region [CDR] loops in gray color). Neighboring residues in FIXa are shown in yellow or red as backbone stick representations with side chains included for residues 338 to 341 and 354. The relative impact of each residue on human FIX binding was evaluated by surface plasmon resonance analysis following amino acid replacement as indicated (right panel) and with the most important residues colored red. Results are shown as residual binding (mean ± standard deviation, n = 9). For all panels, the EGR-chloromethylketone inhibitor (light gray) and calcium ions (light green) are displayed as sphere representations.
As shown in Figure 2B, the anti-FX Fab recognizes a 370-Å2 epitope on FX distributed between the EGF2 (40%) and protease domain (60%). As for the other Mim8 arm, the anti-FX Fab leaves the active site accessible to substrates and inhibitors and, based on available structural models, is predicted to not interfere with the prothrombinase assembly.31,32
The activity of Mim8 is dependent on the stimulatory activity of the anti-FIXa arm
A full titration of FIXa with the Mim8 anti-FIXa OA revealed a (23 ± 0.4) × 103-fold stimulation of FX activation at saturation. This is a 5700-fold enhancement, compared with the parental (biAb46376) anti-FIXa arm, and 31 times higher than that of the emicizumab SIA anti-FIXa OA (Figure 3A). Given the substantially different activities of Mim8 and biAb46376 in HA plasma (Figure 1E), stimulation of the proteolytic activity of FIXa by the anti-FIXa arm thus seems to make a considerable contribution to the overall activity of Mim8.

Mechanisms contributing to the cofactor activity of Mim8. (A) Concentration-dependent stimulation of FX (25 nM) activation by monovalent anti-FIXa OA antibodies (as indicated) in the presence of 0.02 to 2 nM FIXa and 500 µM PS:PC vesicles at pH 7.4. FIXa stimulation (mean ± standard deviation, n = 3) was calculated as the ratio of FX activation in the presence and absence of antibody and fitted to a 1:1 binding model (solid lines) to obtain stimulation indices at saturation of (23 ± 0.4) × 103 and 3.3 ± 0.16 for Mim8 and biAb46376, respectively. In comparison, emicizumab SIA stimulated FX activation 729 ± 11.3–fold relative to free FIXa. (B) FX (25 nM) activation by FIXa (varying concentrations) in the presence of a limiting concentration of Mim8 or emicizumab SIA (0.1 nM) and PS:PC vesicles as indicated (mean ± standard deviation, n = 3). Curve fitting was performed as described in the supplemental Methods to derive a functional KD value of 16.0 ± 2.9 nM for the Mim8-FIXa-FX complex assembly in the presence of membrane. A functional KD value of 0.4 ± 0.1 nM was observed for emicizumab SIA.
Mechanisms contributing to the cofactor activity of Mim8. (A) Concentration-dependent stimulation of FX (25 nM) activation by monovalent anti-FIXa OA antibodies (as indicated) in the presence of 0.02 to 2 nM FIXa and 500 µM PS:PC vesicles at pH 7.4. FIXa stimulation (mean ± standard deviation, n = 3) was calculated as the ratio of FX activation in the presence and absence of antibody and fitted to a 1:1 binding model (solid lines) to obtain stimulation indices at saturation of (23 ± 0.4) × 103 and 3.3 ± 0.16 for Mim8 and biAb46376, respectively. In comparison, emicizumab SIA stimulated FX activation 729 ± 11.3–fold relative to free FIXa. (B) FX (25 nM) activation by FIXa (varying concentrations) in the presence of a limiting concentration of Mim8 or emicizumab SIA (0.1 nM) and PS:PC vesicles as indicated (mean ± standard deviation, n = 3). Curve fitting was performed as described in the supplemental Methods to derive a functional KD value of 16.0 ± 2.9 nM for the Mim8-FIXa-FX complex assembly in the presence of membrane. A functional KD value of 0.4 ± 0.1 nM was observed for emicizumab SIA.
Mim8 preferentially assembles with FIXa and FX on the phospholipid membrane
The interaction of Mim8 with its targets was characterized by ITC, which provided KD values for FIX(a) and FX(a) binding of 4.7 ± 0.7 (2.3 ± 0.3) and 1.5 ± 0.4 (0.3 ± 0.1) µM, respectively (activated forms in parentheses) (supplemental Figures 2 and 3; supplemental Table 2). All titrations were performed at pH 7.4 and 37°C, except for FXa, for which the temperature had to be lowered to 25°C to obtain a measurable endothermic signal. The peak activity of Mim8 concentrations much below these solution-phase KD values (Figure 1E) suggested an effect of membrane on complex assembly. To explore this theory, titrations were performed in the presence of FX, phospholipid, and limiting Mim8, which produced a saturable increase in FX activation as the concentration of FIXa was increased. Because the omission of membrane resulted in greatly reduced FX turnover, it could be concluded that the binding isotherm represented a quaternary Mim8-FIXa-FX-membrane formation for which a global assembly KD of 16 ± 2.9 nM was estimated. In comparison, an assembly KD of 0.4 ± 0.1 nM was determined for emicizumab SIA (Figure 3B).
Activity of Mim8 in plasma- and whole blood–based assays
The ability of Mim8 to promote thrombin generation was tested in human plasma using either FXIa or TF as a trigger to address the intrinsic and extrinsic pathway, respectively. To mimic the human physiological conditions as closely as possible, the concentration of FXIa trigger in congenital HA plasma was optimized to place the dynamic part of the dose response with FVIII in the range of 0 to 100 IU/dL (Figure 4A). Under these conditions, Mim8 provided a maximum level of thrombin generation exceeding that of emicizumab SIA and with a 13-fold improved potency (50% effective concentration) (Figure 4B). With TF as the activator in FVIII-neutralized platelet-rich plasma, normalization was reached at 156 nM Mim8 and with a 50% effective concentration of 23 ± 4.5 nM. Above 625 nM, thrombin generation declined, indicating lower activity due to the hook effect under these conditions. In comparison, in this assay, emicizumab SIA failed to normalize thrombin generation at the highest tested concentration of 4000 nM (Figure 4C). A subsequent comparison of emicizumab SIA to emicizumab revealed a highly similar activity for both molecules (supplemental Figure 5).

Activity of Mim8 in HA plasma and whole blood. (A-B) Peak thrombin generation (mean ± standard deviation, n = 5-8) in FXIa-triggered (1 mU/mL) platelet-poor severe congenital HA plasma supplemented with FVIII (A) or Mim8, emicizumab SIA, or Mim8 in combination with 100 U/dL FVIII (B). Using a variable slope 4-parameter model, 50% effective concentration of 12 ± 2 nM (Mim8) and 153 ± 29 nM (emicizumab SIA) were estimated. (C) Peak thrombin generation of Mim8 or emicizumab SIA in platelet-rich plasma (250 000-320 000 platelets/µL) from healthy volunteers supplemented with neutralizing anti-FVIII polyclonal antibody and triggered with 1 pM TF. (D) Effect of Mim8 or emicizumab SIA on the time to clot (R-time; mean ± standard deviation, n = 6) in whole blood from healthy volunteers supplemented with neutralizing anti-FVIII polyclonal antibody. Clot formation was triggered with TF (∼30 fM) and monitored by using thromboelastography (TEG). Using the same model as in panels A and B, 50% effective concentrations of 0.18 ± 0.02 nM (Mim8) and 6.2 ± 0.9 nM (emicizumab SIA) were estimated. Ranges obtained under normal or HA conditions are highlighted in light blue.
Activity of Mim8 in HA plasma and whole blood. (A-B) Peak thrombin generation (mean ± standard deviation, n = 5-8) in FXIa-triggered (1 mU/mL) platelet-poor severe congenital HA plasma supplemented with FVIII (A) or Mim8, emicizumab SIA, or Mim8 in combination with 100 U/dL FVIII (B). Using a variable slope 4-parameter model, 50% effective concentration of 12 ± 2 nM (Mim8) and 153 ± 29 nM (emicizumab SIA) were estimated. (C) Peak thrombin generation of Mim8 or emicizumab SIA in platelet-rich plasma (250 000-320 000 platelets/µL) from healthy volunteers supplemented with neutralizing anti-FVIII polyclonal antibody and triggered with 1 pM TF. (D) Effect of Mim8 or emicizumab SIA on the time to clot (R-time; mean ± standard deviation, n = 6) in whole blood from healthy volunteers supplemented with neutralizing anti-FVIII polyclonal antibody. Clot formation was triggered with TF (∼30 fM) and monitored by using thromboelastography (TEG). Using the same model as in panels A and B, 50% effective concentrations of 0.18 ± 0.02 nM (Mim8) and 6.2 ± 0.9 nM (emicizumab SIA) were estimated. Ranges obtained under normal or HA conditions are highlighted in light blue.
In HA-induced whole blood initiated by TF, Mim8 shortened the clot time (R-time) and increased the clot development rate (α-angle). At ≥10 nM Mim8, both parameters were close to or within the normal range. In agreement with the results obtained in HA plasma, the potency of Mim8 based on R-time was improved 18-fold, relative to emicizumab SIA (Figure 4D).
Mim8 and FVIII concurrently restore thrombin generation in HA plasma
Structural analysis of Mim8 revealed a recognition site in FIXa known to also be engaged by FVIIIa,33,34 and we therefore explored whether Mim8 would affect plasma procoagulant activity in the presence of exogenous FVIII. Titrations were performed in HA plasma using FXIa as a trigger and with a fixed normalizing concentration (100 U/dL) of FVIII. As shown in Figure 4B, Mim8 caused only a modest dose-dependent reduction in thrombin generation, indicating that plasma procoagulant activity could be restored by FVIII in a wide concentration interval of Mim8.
Mim8 restores hemostasis in HA animal models
In vivo efficacy was tested in HA mouse models of moderate (TVT) and severe (tail clip) bleeding. In the TVT model, it has previously been shown that FVIII administration reduces bleeding to the same level observed for wild-type mice.24 In the tail clip model, a moderate response was observed when FVIII was administered at 14 IU/dL, whereas 59 IU/dL normalized bleeding (Figure 5C). Thus, in both models, administration of FVIII was able to fully restore hemostasis. To overcome the lack of mouse cross-species reactivity of Mim8, HA mice were supplemented with human FIX and FX before the bleeding experiments (Figure 5A).
![Hemostatic effect of Mim8 and emicizumab SIA in HA mouse models of moderate and severe bleeding. (A) Outline of bleeding studies in HA mice after TVT or tail clip. Before vascular injury, mice received sequential IV administrations of human FIX (1.5 mg/kg), human FX (0.9 mg/kg), and Mim8 or emicizumab SIA at indicated doses. (B) Blood loss (mean ± standard error of the mean [SEM], n = 3-6) after TVT. At a given dose, plasma exposure levels of Mim8 and emicizumab SIA were similar, with mean levels (nM) provided on a separate X-axis. The bleeding ranges in vehicle-treated HA and normal animals, respectively, are marked by light blue bars. Applying a 3-parameter inverse log(dose) response model, 50% effective doses of 0.06 mg/kg [0.04-0.14] (Mim8) and 0.7 mg/kg [0.5-1.5] (emicizumab SIA) were estimated (95% confidence intervals in square brackets). (C) Blood loss (mean ± SEM, n = 12; n = 16 for HA control group) following tail clipping. The mean plasma exposure level (nM) of biAb is shown below each dose. Bars on the right depict the blood loss (mean ± SEM, n = 12) in HA mice after administration of three IV doses of FVIII in the absence of supplemented human FIX and FX. Measured plasma levels of FVIII (mean ± SEM) are indicated. Groups were compared with vehicle by using one-way analysis of variance with Dunnett’s multiple comparison test. In addition, groups receiving emicizumab SIA were compared with Mim8 groups receiving the same dose by using a Student t test (2-way, unpaired). P < .05 was considered statistically significant (*P < .05; **P < .01). n.s., not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/138/14/10.1182_blood.2020010331/4/m_bloodbld2020010331f5.png?Expires=1762521959&Signature=iKai-0EdOgqAGMcSr6eGebWtr5Ku5Ro7XhqcJBBR8y8ZGtMcoBfjnmfAoT3bk0TpQTPCv2Egu1dctkp852dLel537Y~N8OV68nxG1RIXzG2TIlYNwqDU1OMBK2NBEU7lRDgbt4moa1MazwfQbQzW-Dh4dqGF1CvQ5BlCqAu9Gq7XOFGievxJryXo7qRLY3T60tNisKvQST0NGbVN~FDhs~7aov7XsndFRwdPvnSUzWjQZnH7t6qATDii~fJV-8azm81NlCMe-yZuhomI0XYY9ZESxV~Ss~B2VUFb~uWoNsK4KFHKM1dy7sdwyfLBwe8p91wE9-WGevG2SjveCsWT8w__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Hemostatic effect of Mim8 and emicizumab SIA in HA mouse models of moderate and severe bleeding. (A) Outline of bleeding studies in HA mice after TVT or tail clip. Before vascular injury, mice received sequential IV administrations of human FIX (1.5 mg/kg), human FX (0.9 mg/kg), and Mim8 or emicizumab SIA at indicated doses. (B) Blood loss (mean ± standard error of the mean [SEM], n = 3-6) after TVT. At a given dose, plasma exposure levels of Mim8 and emicizumab SIA were similar, with mean levels (nM) provided on a separate X-axis. The bleeding ranges in vehicle-treated HA and normal animals, respectively, are marked by light blue bars. Applying a 3-parameter inverse log(dose) response model, 50% effective doses of 0.06 mg/kg [0.04-0.14] (Mim8) and 0.7 mg/kg [0.5-1.5] (emicizumab SIA) were estimated (95% confidence intervals in square brackets). (C) Blood loss (mean ± SEM, n = 12; n = 16 for HA control group) following tail clipping. The mean plasma exposure level (nM) of biAb is shown below each dose. Bars on the right depict the blood loss (mean ± SEM, n = 12) in HA mice after administration of three IV doses of FVIII in the absence of supplemented human FIX and FX. Measured plasma levels of FVIII (mean ± SEM) are indicated. Groups were compared with vehicle by using one-way analysis of variance with Dunnett’s multiple comparison test. In addition, groups receiving emicizumab SIA were compared with Mim8 groups receiving the same dose by using a Student t test (2-way, unpaired). P < .05 was considered statistically significant (*P < .05; **P < .01). n.s., not significant.
Hemostatic effect of Mim8 and emicizumab SIA in HA mouse models of moderate and severe bleeding. (A) Outline of bleeding studies in HA mice after TVT or tail clip. Before vascular injury, mice received sequential IV administrations of human FIX (1.5 mg/kg), human FX (0.9 mg/kg), and Mim8 or emicizumab SIA at indicated doses. (B) Blood loss (mean ± standard error of the mean [SEM], n = 3-6) after TVT. At a given dose, plasma exposure levels of Mim8 and emicizumab SIA were similar, with mean levels (nM) provided on a separate X-axis. The bleeding ranges in vehicle-treated HA and normal animals, respectively, are marked by light blue bars. Applying a 3-parameter inverse log(dose) response model, 50% effective doses of 0.06 mg/kg [0.04-0.14] (Mim8) and 0.7 mg/kg [0.5-1.5] (emicizumab SIA) were estimated (95% confidence intervals in square brackets). (C) Blood loss (mean ± SEM, n = 12; n = 16 for HA control group) following tail clipping. The mean plasma exposure level (nM) of biAb is shown below each dose. Bars on the right depict the blood loss (mean ± SEM, n = 12) in HA mice after administration of three IV doses of FVIII in the absence of supplemented human FIX and FX. Measured plasma levels of FVIII (mean ± SEM) are indicated. Groups were compared with vehicle by using one-way analysis of variance with Dunnett’s multiple comparison test. In addition, groups receiving emicizumab SIA were compared with Mim8 groups receiving the same dose by using a Student t test (2-way, unpaired). P < .05 was considered statistically significant (*P < .05; **P < .01). n.s., not significant.
In the TVT model, dose-response studies showed a significant reduction in blood loss, reaching levels observed in wild-type mice at ≥0.1 mg/kg Mim8 and ≥10 mg/kg emicizumab SIA, corresponding to measured plasma levels ≥10 nM and ≥300 nM, respectively. This difference was mirrored in the estimated 50% effective doses exhibiting a 12-fold difference between Mim8 and emicizumab (Figure 5B). In the tail clip model, a progressive reduction in blood loss was observed from 0.1 to 10 mg/kg Mim8, with the 10 mg/kg dose resulting in a significant blood loss reduction compared with vehicle (Figure 5C). Interestingly, the highest tested dose of 22 mg/kg was less efficacious. With a measured plasma exposure of 2486 nM, we speculate that this is caused by the hook effect. In this model of severe bleeding, emicizumab SIA did not exhibit any significant blood loss reduction at the 3 tested doses of 4.6, 10, and 22 mg/kg.
Pharmacokinetic parameters in cynomolgus monkeys
With affinities of Mim8 for cynomolgus monkey FIX and FX comparable to those measured for the human factors (supplemental Figure 4), this species was chosen for pharmacokinetic studies. The study included 4 different IV doses (0.3, 1.0, 3.0, and 6.0 mg/kg) distributed between 2 arms and 2 dosing occasions, and 1 SC administration performed at 1 mg/kg (Figure 6, top panel). Estimated pharmacokinetic parameters indicate dose linearity in the tested interval and comparable terminal half-lives (12-14 days) and mean residence times (16-20 days) for the 2 administration routes (supplemental Table 3). The data revealed an absorption half-life of 15 hours and a bioavailability of 97% after SC administration, indicating a fast and almost complete absorption of Mim8 in the cynomolgus monkey. No target-mediated drug disposition of Mim8, nor build-up of endogenous FIX or FX, was observed with any of the doses tested (Figure 6).

Pharmacokinetic variables of Mim8 in the cynomolgus monkey. Plasma exposure levels (mean ± standard deviation, n = 4 and 6) of Mim8 and endogenous FIX and FX after IV (left panel) or single-dose (1 mg/kg) SC (right panel) administration, respectively, are shown. For the IV route, 4 dose levels were explored. These were divided between 2 arms and 2 dosing occasions as indicated. Times of administration are marked by dashed vertical lines.
Pharmacokinetic variables of Mim8 in the cynomolgus monkey. Plasma exposure levels (mean ± standard deviation, n = 4 and 6) of Mim8 and endogenous FIX and FX after IV (left panel) or single-dose (1 mg/kg) SC (right panel) administration, respectively, are shown. For the IV route, 4 dose levels were explored. These were divided between 2 arms and 2 dosing occasions as indicated. Times of administration are marked by dashed vertical lines.
Discussion
The molecular design of Mim8 focused on 3 key mechanisms defining the cofactor activity and regulation of FVIII(a). The first mechanism involves the ability of FVIIIa to efficiently assemble with FIXa and FX on the procoagulant membrane surface while interacting minimally with the 2 factors in circulation. With KD values in the low micromolar range for binding of FIX and FX in solution, <0.2% of Mim8 is estimated to engage both factors at relevant plasma levels <350 nM. Consistent with this, a normal antibody half-life was observed in cynomolgus monkeys, with no indications of target mediated drug disposition of Mim8 or accumulation of endogenous FIX and FX. Conversely, inclusion of procoagulant membrane efficiently promoted productive Mim8-FIXa-FX assembly with an apparent affinity that was orders of magnitude stronger than the individual affinities in solution. The most likely explanation for this is a bridging mechanism, mediated by Mim8, in which the 2 coagulation factors are bound in orientations that allow them both to engage the membrane through their N-terminal Gla domains.35,36 Considering that the emicizumab SIA assembled even more tightly on the membrane surface despite solution phase affinities comparable to Mim8 suggests that a considerable orientation element is involved for efficient assembly, as would be expected. A conceptually similar mechanism has been shown for bivalent mAb targeting of high-density surface receptors.37,38 Thus, although the standard biAb format does not incorporate a proteolytic switch as seen for FVIIIa, simultaneous engagement of FIXa and FX seems to provide enough avidity for efficient membrane-localized engagement to occur to recapitulate this property.
The second aspect of the cofactor activity mechanism of FVIIIa that the biAb assembly should be able to provide is facilitation of productive FIXa–FX interactions. This property is dependent on epitope location and affinities, a suboptimal combination of which may reduce activity or enhance the hook effect.21,26 Even at low concentrations, the activity of the high-affinity parental biAb46376 seemed to be compromised by this effect, whereas the mutation-optimized Mim8 exhibited a steady increase in activity up to ∼600 to 1200 nM depending on study conditions.
A third key mechanism of cofactor activity of FVIIIa is allosteric activation of FIXa.16-19 Mechanistic studies suggest that conformational changes are propagated from the cofactor binding site in the 160-helix/170-loop region to the active site where “unlocking” of surrounding loops facilitate efficient substrate turnover.19,33,39 During rounds of mutagenesis, we found that stimulatory activity could be gradually introduced into the anti-FIXa arm of Mim8 and that this was a major contributor to activity in the biAb format, although without providing sufficient procoagulant activity on its own (supplemental Figure 6). The observation that stimulation was solely dependent on the isolated anti-FIXa arm excluded a mechanism driven by standard antibody bivalency. Interestingly, significant stimulatory activity was also observed with the emicizumab SIA anti-FIXa arm, although substantially less than observed for the Mim8 anti-FIXa arm. Considering that this arm recognizes a different epitope on FIXa,21 it may suggest the existence of multiple mechanisms by which the proteolytic activity of FIXa can be enhanced. Anti-FIX antibodies capable of enhancing FIXa-catalyzed FX activation have also been reported previously, with modest rate enhancements in purified membrane-containing systems of ∼10-fold.40 However, because these were tested in the bivalent mAb format and exhibited bell-shaped concentration dependencies, an avidity-driven mechanism relying on improved FIXa localization to the membrane surface cannot be excluded. The molecular details of the mechanism of stimulation are currently an area of investigation.
The efficient FX activation mediated by Mim8 in assays with purified components was reflected by robust thrombin generation and clot formation in plasma- and whole blood–based assays, respectively. Potency of Mim8 was 13 to 18 times higher than that of the emicizumab SIA. Despite an overlapping recognition site with FVIIIa on FIXa, titration of Mim8 into HA plasma supplemented with normal levels of FVIII only modestly affected thrombin generation at the highest concentrations. These results suggest that, if needed, administration of FVIII during Mim8 therapy will achieve a normal procoagulant state.
Assessment of the hemostatic activity of Mim8 in vivo is hampered by the lack of cross-reactivity to FIX and FX from relevant nonclinical species, except non-human primates. To circumvent this issue and enable the use of established bleeding models in HA mice, animals were codosed with human FIX and FX similar to the dosing regimen recently used by Ferrière et al.41 In both moderate (TVT) and severe (tail-clip) challenges, a dose-dependent reduction of blood loss was observed with Mim8. In the moderate challenge model, the plasma concentration–response profile closely followed that obtained in human plasma–based assays, and both Mim8 and emicizumab SIA normalized bleeding but with a 12-fold potency difference in favor of Mim8. In the severe challenge model, additional greater efficacy of Mim8 was illustrated by significantly reduced blood loss, which was not observed for emicizumab SIA. The potency observed for Mim8 indicates that an effective dose for humans could be contained within a small volume (<1 mL). These results, taken with a predictable pharmacokinetic profile, indications of dose linearity, and the observation that SC dosing results in fast and near-complete absorption with good bioavailability, support Mim8 as a next-generation FVIIIa-mimetic candidate, and it is currently undergoing clinical evaluation.
Acknowledgments
The authors thank the entire Novo Nordisk project team for contributions to the development of Mim8, in particular, Mikkel Harndahl, Kristoffer W. Balling, Zhou Jian, and Qiansheng Ren for establishment of high-throughput screen methods, protein purification, and characterization; Anders Svensson and Jens Breinholt for structural investigations; Hermann Pelzer, Esther Bloem, and Laurent Pierre Burnier for functional characterization; Heidi Lindgreen Holmberg for animal studies; and Per Jensen Mikkelsen and Mirella Ezban for scientific advice. The authors also thank Genmab for advice on the DuoBody technology platform used for generation of bispecific antibodies and feedback on the manuscript.
This study was funded by Novo Nordisk A/S. Administrative and editorial support was provided by AXON Communications, funded by Novo Nordisk A/S.
Authorship
Contribution : H.Ø., J.L., P.J.G., N.L., A.H., E.J., G.R., M.G.R., L.B.J., T.E., S.L., H.R.-N., P.S.G., K.L., M.L., L.M.A., A.C.B., F.S., D.E.M., X.L., T.L.H., P.T., H.Z., R.Z., and J.R.B. performed the research; M.G.R., S.L., M.L., H.Z., R.Z., K.T., J.R.B., and B.G.H. contributed new reagents and analytical tools; H.Ø., J.L., C.D.L., and S.K. collected data; H.Ø., J.L., P.J.G., S.K., N.L., A.H., E.J., G.R., M.G.R., L.B.J., T.E., S.L., H.R.N., P.S.G., K.L., M.L., L.M.A., A.C.B., F.S., D.E.M., X.L., T.L.H., C.D.L., P.T., H.Z., R.Z., Z.Y., K.T., M.B.H., J.R.B., B.G.H., and I.H. analyzed and interpreted data; H.Ø., J.L., and S.K. wrote the manuscript; and all authors contributed to the design of the research project.
Conflict-of-interest disclosure: All authors are either current employees of Novo Nordisk A/S, Denmark, or were employees at the time contributing to work in the study. H.Ø., J.L., P.J.G., S.K., N.L., A.H., E.J., M.G.R., L.B.J., S.L., H.R.-N., P.S.G., K.L., M.L., L.M.A., T.L.H., C.D.L., P.T., Z.Y., J.R.B., and B.G.H. are shareholders in Novo Nordisk A/S. H.Ø., P.J.G., E.J., M.G.R., L.B.J., H.Z., R.Z., K.T., Z.Y., and B.G.H. are listed as inventors on pending patents covering Mim8. The remaining authors declare no competing financial interests.
Correspondence: Jacob Lund, Global Drug Discovery, Novo Nordisk A/S, DK-2760 Måløv, Denmark; e-mail: jcld@novonordisk.com; and Per J. Greisen, Global Research Technologies, Novo Nordisk A/S, 2760 Måløv, Denmark; e-mail: pjug@novonordisk.com.
Requests for original data may be submitted to the corresponding authors by e-mail.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal