

Key Points
Expression of target molecules for pediatric AML immunotherapy differs from expression profiles in adult AML.
CD33/CLEC12A are preferential generic pediatric AML combinatorial immunotargets, and CD33/FLT3 are specific to KMT2A-mutated infant AML.

Abstract
Emerging immunotherapies such as chimeric antigen receptor T cells have advanced the treatment of acute lymphoblastic leukemia. In contrast, long-term control of acute myeloid leukemia (AML) cannot be achieved by single lineage-specific targeting while sparing benign hematopoiesis. In addition, heterogeneity of AML warrants combinatorial targeting, and several suitable immunotargets (HAVCR2/CD33 and HAVCR2/CLEC12A) have been identified in adult AML. However, clinical and biologic characteristics of AML differ between children and the elderly. Here, we analyzed 36 bone marrow (BM) samples of pediatric AML patients and 13 age-matched healthy donors using whole RNA sequencing of sorted CD45dim and CD34+CD38−CD45dim BM populations and flow cytometry for surface expression of putative target antigens. Pediatric AML clusters apart from healthy myeloid BM precursors in principal-component analysis. Known immunotargets of adult AML, such as IL3RA, were not overexpressed in pediatric AML compared with healthy precursors by RNA sequencing. CD33 and CLEC12A were the most upregulated immunotargets on the RNA level and showed the highest surface expression on AML detected by flow cytometry. KMT2A-mutated infant AML clusters separately by RNA sequencing and overexpresses FLT3, and hence, CD33/FLT3 cotargeting is an additional specific option for this subgroup. CLEC12A and CD33/CLEC12Adouble-positive expression was absent in CD34+CD38−CD45RA−CD90+ hematopoietic stem cells (HSCs) and nonhematopoietic tissue, while CD33 and FLT3 are expressed on HSCs. In summary, we show that expression of immunotargets in pediatric AML differs from known expression profiles in adult AML. We identify CLEC12A and CD33 as preferential generic combinatorial immunotargets in pediatric AML and CD33 and FLT3 as immunotargets specific for KMT2A-mutated infant AML.
Introduction
Therapy of acute myeloid leukemia (AML) has recently improved by optimization of chemotherapy regimens, risk stratification, and supportive care.1,2 However, both pediatric and adult AML patients still have a dismal prognosis and have not benefited from recent advances in the field of immunotherapy. On the contrary, patients with B-cell precursor acute lymphoblastic leukemia have benefited from recent advances (eg, bispecific T-cell engager blinatumomab and CD19-directed chimeric antigen receptor [CAR] T cells).3-6 In recent years, several immunotherapy approaches have been investigated in patients with AML.7 Redirecting the patient’s own immune system to target cancer cells using bispecific T-cell engaging antibodies, CAR T cells, or CAR natural killer cells is a highly attractive approach that aims to simultaneously enhance antitumor activity while reducing the burden of systemic toxicities seen with cytotoxic chemotherapy.8 As pediatric AML patients suffer from long-term side effects of intensive chemotherapy regimens, there is an urgent need for new and less toxic therapies. However, target antigen selection for immunotherapy has remained a challenge in both pediatric and adult AML. Selection of a lineage-specific marker as a target for immunotherapy could lead to an elimination of healthy myeloid hematopoiesis, resulting in life-threatening neutropenia. Consequently, immunotherapy for AML can warrant a “bridge-to-transplant” strategy, where the utmost aim is to eliminate AML prior to allogeneic stem cell transplantation. No ideal single target molecule has been identified yet for AML. Recent efforts focus on combinatorial targeting in order to achieve 2 goals: (1) increase the percentage of AML cells targeted and (2) reduce on-target off-leukemia toxicity.9,10
CD33 and CD123 (IL3RA) are the antigens most frequently explored in clinical trials as single targets for bispecific antibodies and CARs in AML.11 CD33 is a transmembrane sialic acid–binding immunoglobulin-type lectin receptor expressed on the surface of AML cells.12 CD33 is expressed on mature myeloid cells and hematopoietic progenitor cells, which must be taken into account for expected on-target off-tumor toxicity. CD123 is a glycoprotein of the α subunit of the interleukin-3 receptor and is broadly expressed on adult AML.13,14 Beyond CD33 and IL3RA, C-type lectin-like receptor 1 (also known as CLEC12A) is highly expressed in AML and shows absent or low expression on hematopoietic stem cells (HSCs) but is present on mature myeloid cells.15-17 Moreover, CD135, also known as FMS-like tyrosine kinase 3 (FLT3), is part of the receptor tyrosine kinase class III family and is widely expressed not only on AML blasts but also on early and late physiologic hematopoietic progenitors.18,19
In order to increase target specificity and still cover heterogeneity of AML, differentially expressed targets have been investigated using integrated screening efforts in order to determine candidate antigen combinations for targeted immunotherapy in adult AML.9 CD33/HAVCR2 and CLEC12A/HAVCR2 have been recently identified as preferential combinatorial immunotherapy targets.10 While the distinct genomic landscape of pediatric AML is well known,11,20 systematic analysis of combinatorial target antigen expression patterns for synthetic immunotherapy is still missing. Consequently, we analyzed a well-defined cohort of therapy-naive bone marrow (BM) aspirates from pediatric AML patients at time of diagnosis and additionally healthy age-matched individuals by both RNA sequencing of sorted CD45dim and CD45dimCD34+CD38− cells and subsequent flow cytometry of potential surface antigens (CD33, CLEC12A, CD38, IL3RA, FLT3, HAVCR2, EMR2, LILRB2, CD96, CD70, and CCR1) in order to identify target combinations specific for pediatric AML. The goal of this study is to pave the way for synthetic immunotherapy development for pediatric AML.
Patients, materials, and methods
Patients
Patient and healthy donor characteristics are shown in Table 1.21 This study was approved by the Institutional Ethical Review Board (“Ethikkommission bei der LMU München," approval number 17-163) and performed in accordance with the Declaration of Helsinki. Patients and healthy donors or their legal guardians gave written informed consent or analysis was performed from anonymized samples from the local biobank. BM samples from AML patients and healthy BM donors were kindly provided by the Hauner Hematology Biobank. Healthy individuals are age-matched children without any evidence of hematological disease. Most patients contributed 1 sample to this study, but 3 patients contributed 2 samples each (primary and relapse) and 1 patient contributed 4 samples (several relapses). KMT2A aberrations consisted of KMT2A fusions to AF9 (twice), AF10 (once), and ENL (once) and no fusion partner for KMT2A documented in 1 patient sample.
Characteristics of AML patients and healthy controls
| Initial samples (n = 21) | Relapse samples (n = 15) | Healthy BM donors (n = 13) | |
|---|---|---|---|
| Age (y), mean (range) | 7.5 (0.4-16.6) | 8.5 (1.9-17.1) | 8.8 (1.7-19.1) |
| Gender (male/female) | 11/10 | 6/9 | 4/9 |
| Age at initial diagnosis (y), mean (range) | 7.5 (0.4-16.6) | 6.3 (0.4-16.5) | — |
| AML blasts frequency by flow cytometry (%) | 68 (40-96) | 51 (20-80) | — |
| Future relapse, n (%) | 7 (33.3) | — | |
| Time to relapse (y), mean (range) | 1.2 (0.2-1.8) | — | |
| Death, n (%) | 3 (14.3) | 7 (46.7) | — |
| Monitoring period (y), mean (range) | 7.0 (0.8-14.4) | 7.3 (0.9-14.4) | — |
| FAB phenotype, n (%) | |||
| M0 | 1 (4.8) | 1 (6.7) | — |
| M1 | 2 (9.5) | 0 (0) | — |
| M2 | 5 (23.8) | 3 (20.0) | — |
| M3 | 0 (0) | 0 (0) | — |
| M4 | 6 (28.6) | 4 (26.7) | — |
| M4eo | 1 (4.8) | 2 (13.3) | — |
| M5 | 3 (14.3) | 4 (26.7) | — |
| M6 | 1 (4.8) | 0 (0) | — |
| M7 | 1 (4.8) | 0 (0) | — |
| Unknown | 1 (4.8) | 1 (6.7) | — |
| Cytogenetic aberrations, n (%) | |||
| Normal karyotype | 5 (23.8) | 4 (26.7) | — |
| t(8;21) | 5 (23.8) | 2 (13.3) | — |
| Inv(16) | 0 (0) | 2 (13.3) | — |
| Trisomy 8 | 1 (4.8) | 0 (0) | — |
| Del(9q) | 1 (4.8) | 0 (0) | — |
| t(6;9) | 1 (4.8) | 0 (0) | — |
| Del(17p)/p53 | 1 (4.8) | 0 (0) | — |
| Trisomy 6 + 7 | 1 (4.8) | 0 (0) | — |
| t(7;12)(q36, p13) + trisomy 19 | 0 (0) | 4 (26.7) | — |
| Del(12p) | 0 (0) | 1 (6.7) | — |
| Other abnormalities | 3 (14.3) | 0 (0) | — |
| Unknown | 3 (14.3) | 2 (13.3) | — |
| Mutations, n (%) | |||
| FLT3.ITD | 2 (9.5) | 1 (6.7) | — |
| KMT2A fusion | 4 (19.0) | 1 (6.7) | — |
| Initial samples (n = 21) | Relapse samples (n = 15) | Healthy BM donors (n = 13) | |
|---|---|---|---|
| Age (y), mean (range) | 7.5 (0.4-16.6) | 8.5 (1.9-17.1) | 8.8 (1.7-19.1) |
| Gender (male/female) | 11/10 | 6/9 | 4/9 |
| Age at initial diagnosis (y), mean (range) | 7.5 (0.4-16.6) | 6.3 (0.4-16.5) | — |
| AML blasts frequency by flow cytometry (%) | 68 (40-96) | 51 (20-80) | — |
| Future relapse, n (%) | 7 (33.3) | — | |
| Time to relapse (y), mean (range) | 1.2 (0.2-1.8) | — | |
| Death, n (%) | 3 (14.3) | 7 (46.7) | — |
| Monitoring period (y), mean (range) | 7.0 (0.8-14.4) | 7.3 (0.9-14.4) | — |
| FAB phenotype, n (%) | |||
| M0 | 1 (4.8) | 1 (6.7) | — |
| M1 | 2 (9.5) | 0 (0) | — |
| M2 | 5 (23.8) | 3 (20.0) | — |
| M3 | 0 (0) | 0 (0) | — |
| M4 | 6 (28.6) | 4 (26.7) | — |
| M4eo | 1 (4.8) | 2 (13.3) | — |
| M5 | 3 (14.3) | 4 (26.7) | — |
| M6 | 1 (4.8) | 0 (0) | — |
| M7 | 1 (4.8) | 0 (0) | — |
| Unknown | 1 (4.8) | 1 (6.7) | — |
| Cytogenetic aberrations, n (%) | |||
| Normal karyotype | 5 (23.8) | 4 (26.7) | — |
| t(8;21) | 5 (23.8) | 2 (13.3) | — |
| Inv(16) | 0 (0) | 2 (13.3) | — |
| Trisomy 8 | 1 (4.8) | 0 (0) | — |
| Del(9q) | 1 (4.8) | 0 (0) | — |
| t(6;9) | 1 (4.8) | 0 (0) | — |
| Del(17p)/p53 | 1 (4.8) | 0 (0) | — |
| Trisomy 6 + 7 | 1 (4.8) | 0 (0) | — |
| t(7;12)(q36, p13) + trisomy 19 | 0 (0) | 4 (26.7) | — |
| Del(12p) | 0 (0) | 1 (6.7) | — |
| Other abnormalities | 3 (14.3) | 0 (0) | — |
| Unknown | 3 (14.3) | 2 (13.3) | — |
| Mutations, n (%) | |||
| FLT3.ITD | 2 (9.5) | 1 (6.7) | — |
| KMT2A fusion | 4 (19.0) | 1 (6.7) | — |
FAB, French‐American‐British (Cooperative Group classification).21
Flow cytometry
ll samples measured in this project were subjected to mononuclear cell isolation using density centrifugation (Biocoll, Merck), cryopreservation with 5% human serum albumin (Biotest Pharma), and 20% dimethyl sulfoxide (Sigma Aldrich) and storage in liquid nitrogen. BM cells of AML patients and healthy BM donors were stained with AF488-CCR1 (clone 5F10B29), BV650-CD33 (clone WM53), PE-LILRB2 (clone 42D1), PerCPCy5.5-FLT3 (clone BV10A4H2), BV785-IL3RA (clone 6H6), PerCPCy5.5-CD70 (clone 113-16), PE-FLT3 (clone BV10A4H2), APC-CD90 (clone 5E10), and PerCPCy5.5-CD45RA (clone HI100) from BioLegend (San Diego, CA); BV421-CLEC12A (clone 50C1), AF647-EMR2 (clone 494025), BB515-HAVCR2 (clone 7D3), BUV496-CD38 (clone HIT2), BUV395-CD45 (clone HI30), BB515-CD96 (clone 6F9), PE-CF594-CD45 (clone HI30), and BUV737-CD10 (clone HI10a) from Becton, Dickinson and Company (BD) (Franklin Lakes, NJ); PEVio770-CD34 (clone AC136) and VioBlue-CD45 (clone 5B1) from Miltenyi Biotec (Bergisch Gladbach, Germany); and fixable viability dye eFluor 780 from eBioscience/Thermo Fisher Scientific (Waltham, MA).
Flow cytometry analysis was performed on a BD LSRFortessa, while fluorescence-activated cell sorting (FACS) of CD45dim BM populations was performed on a BD FACSAria III. Flow cytometry data were analyzed using FlowJo X (FlowJo, Ashland, OR). For analysis of surface molecules, either the percentage of positive cells was counted using an isotype control as negative gating control or the median fluorescence intensity (MFI) ratio was calculated by dividing the MFI of the stained sample by the isotype control. tSNE (t-distributed stochastic neighbor embedding) was performed using the integrated tSNE algorithm of FlowJo X. To that end, 18 representative AML samples were used to generate a concatenated flow cytometry standard (.fcs) file using an equal amount of cells per sample to a total of <100 000 in order to facilitate local computing of tSNE parameters. Next, individually compensated parameters (EMR2, CD34, CD38, CD33, IL3RA, FLT3, CLEC12A) were used to calculate tSNE parameters that were used for all graphs in this article. In the tSNE analysis (Figure 6G), samples from healthy donors were included in tSNE parameter calculation, leading to a different map based on CD33, CD34, CD38, CLEC12A IL3RA and FLT3 expression. When expression of CD33, HAVCR2 and CLEC12A was further analyzed on leukemic stem cells (LSCs), we compared expression of these markers on bulk AML and on CD34+CD38− AML cells, which we defined as leukemic stem cells (supplemental Figure 1B, available on the Blood Web site).
For calculation of the MFI ratio, compensated median fluorescence was exported from FlowJo X and subsequently analyzed in a spreadsheet program. Next, MFI of the stained samples was divided by MFI of isotype control sample. As isotype MFI was negative in some cases, we included a stabilizing factor in this calculation, and the final values illustrated in this article were calculated as follows: MFI ratio = (MFI(stained) + 200)/(MFI(isotype) + 200). While this might lead to an underestimation of surface expression, it reduces the noise of the approach and avoids negative MFI ratios.
RNA sequencing
RNA was isolated from sorted BM populations using a Quick-RNA Microprep Kit (Zymo, Irvine, CA). Quality of RNA was determined using the Agilent RNA 6000 Pico Kit on Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA), and only samples with RNA integrity number >7 were sequenced. RNA-sequencing library preparation was done after poly(A) mRNA isolation using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs, Ipswich, MA). We performed Illumina sequencing with 2 × 75-bp paired-end reads on the NextSeq500 sequencer (Illumina, San Diego, CA).
Bioinformatics
Raw sequencing reads were processed using Trim Galore (v0.4.4) (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) and subsequently mapped against the GRCh37.p13 reference genome by applying the RNA-sequencing aligner STAR (v2.5.0a).22 Expression counts were generated using featureCounts (v1.5.2)23 and gene structure information for GRCh37 as specified by Ensembl 87. Principal-component analysis (PCA) was performed after variance-stabilizing transformation using DESeq2. Significance of differential expression of RNA-sequencing data were determined using the DESeq2 package.24 All AML samples were additionally screened for somatic mutations in 49 genes that are relevant to AML diagnosis and prognosis by applying the RNAmut (v1.1) pipeline (for tested genes, see supplemental Table 2).25
Statistics
We determined statistical significance of differential surface marker expression with Prism 8 using the Wilcoxon-Mann-Whitney test, as our sample size was not sufficient to test for normal distribution (GraphPad, San Diego, CA).
Results
Transcriptome of purified pediatric AML blasts reveal overexpression of immunotargets compared with healthy hematopoietic precursors
Before analyzing the transcriptome, primary blasts were purified by FACS from pediatric BM samples. In order to identify expressed surface target molecules in pediatric acute leukemia blasts, we performed RNA sequencing of sorted CD45dim leukemic blast populations from diagnostic BM samples (n = 36) of pediatric AML patients and CD45dim hematopoietic progenitors (n = 13) from healthy control individuals (Table 1; supplemental Figure 1A). We found that pediatric AML samples segregate from healthy BM progenitors by PCA of RNA-sequencing data (Figure 1A). Moreover, we observed clustering of pediatric AML samples according to KMT2A and t(8;21) status. Next, we determined differential expression of genes in pediatric AML samples compared with healthy hematopoietic progenitors. Among the most differentially expressed genes were immunotherapy targets previously reported on adult AML (Figure 1B).9 In particular, CD33, ITGB5, CD244, CLEC12A, P2RY13, CD7, CD70, and CD96 were significantly enriched in pediatric AML samples (Figure 1C). Detailed information on overexpression of reported AML targets or surface molecules can be found in supplemental Tables 1 and 3. In order to determine which genes are overexpressed in AML when compared with early hematopoietic precursors (defined by CD45dimCD34+CD38− sorting from healthy BM), we performed differential RNA-sequencing analysis between AML and 18 healthy early hematopoietic precursor cells (Figure 1D). We observed that CD33, CLEC12A, CD96, P2RY13, CD70, and ITGB5 were overexpressed in AML compared with early hematopoietic precursors (Figure 1E).
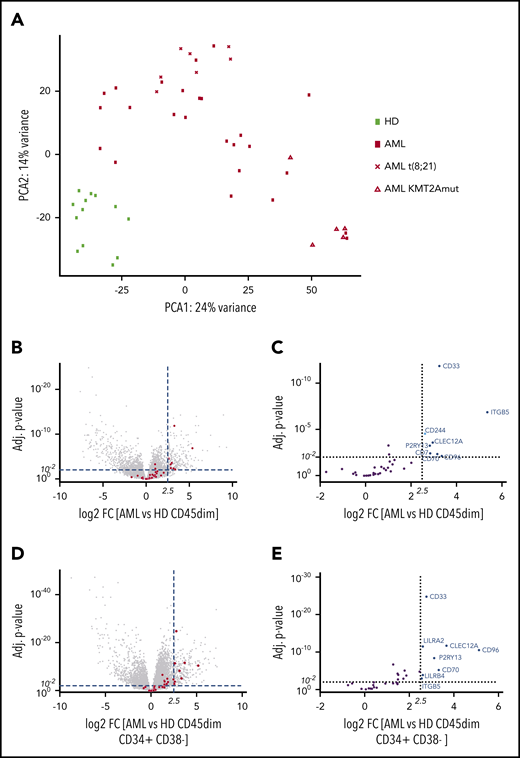
Pediatric AML clusters by RNA sequencing from healthy hematopoietic progenitors and overexpresses AML immunotargets such as CD33 and CLEC12A. (A) PCA of 49 samples by RNA sequencing, including 36 AML samples and 13 healthy hematopoietic progenitors. Samples cluster according to disease status, while t(8;21) and KMT2A-mutated samples form clusters within AML samples. One myelodysplastic syndrome–associated AML sample was excluded from this PCA. (B) Volcano plot of all differentially expressed genes with >100 base mean expression according to DESe2 analysis in healthy CD45dim hematopoietic progenitors and pediatric AML samples. AML immunotargets described by Perna et al9 are depicted by red dots. Dotted lines illustrate cutoff values of log2 fold change of 2.5 (22,5 ∼5.6) and adjusted P < .01 to define significantly highly enriched genes in sorted AML blasts. (C) Volcano plot detailing differential expression of 35 AML immunotargets described by Perna et al in pediatric AML blasts. (D) Volcano plot of all differentially expressed genes with >100 base mean expression according to DESeq2 analysis in healthy CD45dimCD34+CD38− and pediatric AML samples. AML immunotargets described by Perna et al9 are depicted by red dots. Dotted lines illustrate cutoff values of log2 fold change of 2.5 (22,5 ∼5.6) and adjusted P < .01 to define significantly highly enriched genes in sorted AML blasts. (E) Volcano plot detailing differential expression of 35 AML immunotargets described by Perna et al in pediatric AML blasts. FC, fold change; HD, healthy donor; mut, mutated.
Pediatric AML clusters by RNA sequencing from healthy hematopoietic progenitors and overexpresses AML immunotargets such as CD33 and CLEC12A. (A) PCA of 49 samples by RNA sequencing, including 36 AML samples and 13 healthy hematopoietic progenitors. Samples cluster according to disease status, while t(8;21) and KMT2A-mutated samples form clusters within AML samples. One myelodysplastic syndrome–associated AML sample was excluded from this PCA. (B) Volcano plot of all differentially expressed genes with >100 base mean expression according to DESe2 analysis in healthy CD45dim hematopoietic progenitors and pediatric AML samples. AML immunotargets described by Perna et al9 are depicted by red dots. Dotted lines illustrate cutoff values of log2 fold change of 2.5 (22,5 ∼5.6) and adjusted P < .01 to define significantly highly enriched genes in sorted AML blasts. (C) Volcano plot detailing differential expression of 35 AML immunotargets described by Perna et al in pediatric AML blasts. (D) Volcano plot of all differentially expressed genes with >100 base mean expression according to DESeq2 analysis in healthy CD45dimCD34+CD38− and pediatric AML samples. AML immunotargets described by Perna et al9 are depicted by red dots. Dotted lines illustrate cutoff values of log2 fold change of 2.5 (22,5 ∼5.6) and adjusted P < .01 to define significantly highly enriched genes in sorted AML blasts. (E) Volcano plot detailing differential expression of 35 AML immunotargets described by Perna et al in pediatric AML blasts. FC, fold change; HD, healthy donor; mut, mutated.
CD33 and CLEC12A are consistently overexpressed by pediatric AML by flow cytometry
Next, we selected 11 AML target proteins based on literature research and our RNA-sequencing data on enrichment in pediatric AML blasts (CD33, CLEC12A, CD96, CD70, CD38, IL3RA, FLT3, HAVCR2, EMR2, LILRB2, and CCR1) and analyzed surface expression by flow cytometry on AML blasts and healthy hematopoietic progenitors. ITGB5 was excluded because of ubiquitous expression (Figure 2E).26 In order to determine the suitability of target molecules for pediatric AML immunotherapy, we measured both the percentage of AML cells expressing the target and the MFI ratio as a parameter of target density on the surface of AML cells (Figure 2A,C). Only CD33, CLEC12A, and CD38 showed a mean expression on pediatric AML blasts of >50%, and the MFI ratio was highest for CD33 and CLEC12A (Figure 2A,C). When we determined expression of target molecules on healthy hematopoietic progenitors (CD45dim cells), we observed high CD38 expression, while CD33 and CLEC12A showed low expression in this gated population (Figure 2B,D; supplemental Figure 1D).
![CD33 and CLEC12A are highly expressed in pediatric AML and widely absent in both healthy hematopoietic and nonhematopoietic tissue. (A) Percentage of leukemic blasts, defined as CD45dim cells within the initial BM of an AML patient (n = 28-36; see also supplemental Figure 1A), expressing 1 of 11 surface molecules (mean ± standard deviation [SD]). Gating of flow cytometry was performed using isotype control antibodies. (B) Percentage of leukemic blasts and healthy hematopoietic progenitors (CD45dim cells within healthy BM; see supplemental Figure 1A) expressing 1 of 8 selected surface molecules (mean ± SD). (C) MFI ratio of 11 surface molecules on leukemic blasts. Because results are from multicolor flow cytometry, quantitative comparison between target antigens using different fluorochromes allows only limited semiquantitative analysis. (D) MFI ratio of 8 selected surface molecules on pediatric AML blasts and healthy hematopoietic progenitors. (E) Expression of 11 surface proteins analyzed in this study and 4 more genes found to be overexpressed in pediatric AML (CD7, P2RY13, CD244, and ITGB5) in healthy tissues using the ProteomicsDB26,27 repository. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as ubiquitously expressed gene and CD19 as suitable immunotarget for B-cell precursor ALL are depicted in green. Genes are clustered according to similar expression patterns.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/8/10.1182_blood.2020006921/1/m_bloodbld2020006921f2.png?Expires=1769084695&Signature=KGsjX69jtBWc36hpsx4Tcyv0CzHYIHQee6pPgZvo5~cNeD3hqxJEFep0TEAIDL3NFSTTSryhMP6Ikm727GDrwGxeCMdW1~edgzn-Be68Sxp3lQIUXwAFKTDZNgUtSi6SePRfWQHaJeZNag7wa2CJNYObc2rRJq5uWPD36xIMZZcwHQ1eh5mH8k1JJN6xI7KJfe0E~9pDWsn~vMliqJBShLRJ64Mn7EXKTupiDx2v6zmJBP52gJj18jL8v4ZyjBlZhQ08ZkHpR~oDBfYl9~SlLzGMcO-4dniWTVz7GhiRoLeZ8EfytiZ1zX~kvCP4VSdGZJZXSmsFbALBO5EYU5h1aw__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
CD33 and CLEC12A are highly expressed in pediatric AML and widely absent in both healthy hematopoietic and nonhematopoietic tissue. (A) Percentage of leukemic blasts, defined as CD45dim cells within the initial BM of an AML patient (n = 28-36; see also supplemental Figure 1A), expressing 1 of 11 surface molecules (mean ± standard deviation [SD]). Gating of flow cytometry was performed using isotype control antibodies. (B) Percentage of leukemic blasts and healthy hematopoietic progenitors (CD45dim cells within healthy BM; see supplemental Figure 1A) expressing 1 of 8 selected surface molecules (mean ± SD). (C) MFI ratio of 11 surface molecules on leukemic blasts. Because results are from multicolor flow cytometry, quantitative comparison between target antigens using different fluorochromes allows only limited semiquantitative analysis. (D) MFI ratio of 8 selected surface molecules on pediatric AML blasts and healthy hematopoietic progenitors. (E) Expression of 11 surface proteins analyzed in this study and 4 more genes found to be overexpressed in pediatric AML (CD7, P2RY13, CD244, and ITGB5) in healthy tissues using the ProteomicsDB26,27 repository. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as ubiquitously expressed gene and CD19 as suitable immunotarget for B-cell precursor ALL are depicted in green. Genes are clustered according to similar expression patterns.
CD33 and CLEC12A are highly expressed in pediatric AML and widely absent in both healthy hematopoietic and nonhematopoietic tissue. (A) Percentage of leukemic blasts, defined as CD45dim cells within the initial BM of an AML patient (n = 28-36; see also supplemental Figure 1A), expressing 1 of 11 surface molecules (mean ± standard deviation [SD]). Gating of flow cytometry was performed using isotype control antibodies. (B) Percentage of leukemic blasts and healthy hematopoietic progenitors (CD45dim cells within healthy BM; see supplemental Figure 1A) expressing 1 of 8 selected surface molecules (mean ± SD). (C) MFI ratio of 11 surface molecules on leukemic blasts. Because results are from multicolor flow cytometry, quantitative comparison between target antigens using different fluorochromes allows only limited semiquantitative analysis. (D) MFI ratio of 8 selected surface molecules on pediatric AML blasts and healthy hematopoietic progenitors. (E) Expression of 11 surface proteins analyzed in this study and 4 more genes found to be overexpressed in pediatric AML (CD7, P2RY13, CD244, and ITGB5) in healthy tissues using the ProteomicsDB26,27 repository. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as ubiquitously expressed gene and CD19 as suitable immunotarget for B-cell precursor ALL are depicted in green. Genes are clustered according to similar expression patterns.
CD33 and CLEC12A expression is limited to hematopoietic cells, while ITGB5 and CD38 are widely expressed in healthy tissues
Subsequently, we analyzed expression of AML target molecules on healthy tissues using the publicly available repository ProteomeDB.27 As reference, we included glyceraldehyde-3-phosphate dehydrogenase as ubiquitously expressed protein and CD19 as established immunotherapy target into our analysis (Figure 2E). Based on proteome data, CD33 and CLEC12A are coexpressed in myeloid cells, such as monocytes. A weak coexpression is also detected in the lung, which, however, does not exceed CD19 expression in the lung (CD19 CAR T cells have not attacked lung tissue). Furthermore, we found that ITGB5 and CD38 are heavily expressed in a variety of healthy tissues and consequently are not suitable for AML immunotherapy. Several AML immunotargets are expressed on healthy immune cells (monocytes, T cells, B cells, and natural killer cells) or in organs that contain many immune cells (spleen and lymph nodes). In summary, CD33 and CLEC12A, the most highly expressed immunotargets for pediatric AML, have no inferior expression profile in healthy tissue for immunotherapy compared with other target molecules (Figure 2E).
CLEC12A and HAVCR2 are absent on HSCs in healthy children and adults, while CD33 and FLT3 are widely expressed on physiologic early hematopoietic progenitors
Expression of the 4 promising target molecules (CD33, CLEC12A, FLT3, and HAVCR2) was analyzed on physiologic hematopoietic precursors in order to determine the impact of synthetic immunotherapy directed against these molecules. We measured surface protein expression on 7 hematopoietic progenitors subpopulations in pediatric and adult BM (Figure 3A)28-30 : HSCs (CD34+CD38−CD45RA−CD90+), multipotent progenitors (MPPs; CD34+CD38−CD45RA−CD90−), multipotent lymphoid progenitors (MLPs; CD34+CD38−CD45RA+CD90−), common myeloid progenitors (CMPs; CD34+CD38+CD10−CD45RA−FLT3+), megakaryocyte/erythroid progenitors (CD34+CD38+CD10−CD45RA−FLT3−), granulocyte-macrophage progenitors (GMPs; CD34+CD38+CD10−CD45RA+FLT3+), and common lymphoid progenitors (CD34+CD38+CD10+). While FLT3 is highly expressed on both HSCs and more differentiated subsets, CD33 is expressed to a lower extent (eg, 50% in pediatric HSCs) in early hematopoiesis and more highly expressed in differentiated myeloid cells (Figure 3B,D). CLEC12A is not expressed in HSCs but is highly expressed in differentiated myeloid cells (eg, monocytes; Figure 3C). HAVCR2 expression is low in HSCs, with an increasing expression in more differentiated myeloid compartments (supplemental Figure 2I). No principal difference was observed between target antigen expression in pediatric and adult HSCs. Our flow cytometry data are supported by transcriptome data from a publicly available data set (supplemental Figures 1E and 2H). Additionally, pediatric HSCs and adult HSCs cluster together in transcriptome analysis when compared with pediatric CD45dim populations (supplemental Figure 3A-B). FLT3, CD33, CLEC12A, and HAVCR2 expression on HSCs is consistent across single individuals (supplemental Figure 4).
![Expression of CD33, CLEC12A, FLT3, and HAVCR2 in pediatric and adult hematopoietic progenitors. (A) Gating strategy used for definition of 7 different hematopoietic progenitors downstream of gating on cells (side scatter [SSC]-A vs forward scatter [FSC]-A), single cells (FSC-H vs FSC-A), and living cells (SSC-A vs viability dye): MLPs, MPPs, HSCs, common lymphoid progenitors (CLPs), megakaryocyte/erythroid progenitors (MEPs), GMPs, and CMPs. Additionally, monocytes were gated according to SSC-A vs CD45 gating. (B) FLT3 expression by percentage (top) and MFI ratio (bottom). (C) CLEC12A expression by percentage (top) and MFI ratio (bottom). (D) CD33 expression by percentage (top) and MFI ratio (bottom). (E) Percentage of hematopoietic progenitors expressing both CD33 and CLEC12A. (F) Percentage of hematopoietic progenitors expressing both CD33 and FLT3. (G) Percentage of hematopoietic progenitors expressing both CD33 and HAVCR2.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/137/8/10.1182_blood.2020006921/1/m_bloodbld2020006921f3.png?Expires=1769084695&Signature=mWf04ZpNQToWMPm~~ITUe0~XNqRddNAFF~QWnE6JLvqzbMA0qHDkHRlsAkavva7pwNv-MmnQiErzUb31BXKHPdMZ7DHY4VQjnnKxWDZ-g59wg5HLuiUzFMaJJ1MGi8cSKwhDPcmAY1MZoPUlMvT5LDI3ZYhZYeZAcOMwJBML~jbIMUxhp69asBbcuTB7OQk16EaJ400aISWGlH9TQdPV-TffjBaXlYVXllI-MP7N94ZMdRMV8um4edxTZKM0REe3o1R6VyxpoqS5xvZxWTc-aam8JwfS3YaZan8YTSLyVH6is6KGi38od3ssk1zIWD2gPBAMzE8pWGuPcM~qCC2g7Q__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Expression of CD33, CLEC12A, FLT3, and HAVCR2 in pediatric and adult hematopoietic progenitors. (A) Gating strategy used for definition of 7 different hematopoietic progenitors downstream of gating on cells (side scatter [SSC]-A vs forward scatter [FSC]-A), single cells (FSC-H vs FSC-A), and living cells (SSC-A vs viability dye): MLPs, MPPs, HSCs, common lymphoid progenitors (CLPs), megakaryocyte/erythroid progenitors (MEPs), GMPs, and CMPs. Additionally, monocytes were gated according to SSC-A vs CD45 gating. (B) FLT3 expression by percentage (top) and MFI ratio (bottom). (C) CLEC12A expression by percentage (top) and MFI ratio (bottom). (D) CD33 expression by percentage (top) and MFI ratio (bottom). (E) Percentage of hematopoietic progenitors expressing both CD33 and CLEC12A. (F) Percentage of hematopoietic progenitors expressing both CD33 and FLT3. (G) Percentage of hematopoietic progenitors expressing both CD33 and HAVCR2.
Expression of CD33, CLEC12A, FLT3, and HAVCR2 in pediatric and adult hematopoietic progenitors. (A) Gating strategy used for definition of 7 different hematopoietic progenitors downstream of gating on cells (side scatter [SSC]-A vs forward scatter [FSC]-A), single cells (FSC-H vs FSC-A), and living cells (SSC-A vs viability dye): MLPs, MPPs, HSCs, common lymphoid progenitors (CLPs), megakaryocyte/erythroid progenitors (MEPs), GMPs, and CMPs. Additionally, monocytes were gated according to SSC-A vs CD45 gating. (B) FLT3 expression by percentage (top) and MFI ratio (bottom). (C) CLEC12A expression by percentage (top) and MFI ratio (bottom). (D) CD33 expression by percentage (top) and MFI ratio (bottom). (E) Percentage of hematopoietic progenitors expressing both CD33 and CLEC12A. (F) Percentage of hematopoietic progenitors expressing both CD33 and FLT3. (G) Percentage of hematopoietic progenitors expressing both CD33 and HAVCR2.
Finally, we analyzed expression of CD33 and CLEC12A in normal blood cells and found coexpression only in monocytes, promyelocytes, and myelocytes using the Web tool bloodspot (supplemental Figure 1E).31 Granulocyte-monocyte progenitors and myeloid dendritic cells expressed only CD33 and showed low levels of CLEC12A.
Heterogeneity of pediatric AML limits specific targeting of LSCs in children
As targeting of LSCs has been discussed as a requirement for AML immunotargeting,10 we first analyzed CD34+CD38− AML subset frequency in our pediatric cohort, as this population has been described to contain the most AML LSCs.32 We observed that a mean of 24% of AML cells were CD34+CD38−, with an extremely wide frequency range (0.9% to 88.4%). Many samples contained a very low frequency of this phenotype, while healthy CD45dim populations had a frequency of 5.1% (range, 2.8% to 7.1%; Figure 4A-B). Consequently, in AML samples with low or very high CD34+CD38− frequency, CD34/CD38 gating allowed no clear definition of LSCs in pediatric AML. In addition, in this pediatric AML cohort, the LSC gate allowed no distinction of LSCs from contaminating healthy hematopoietic progenitors with the same phenotype.
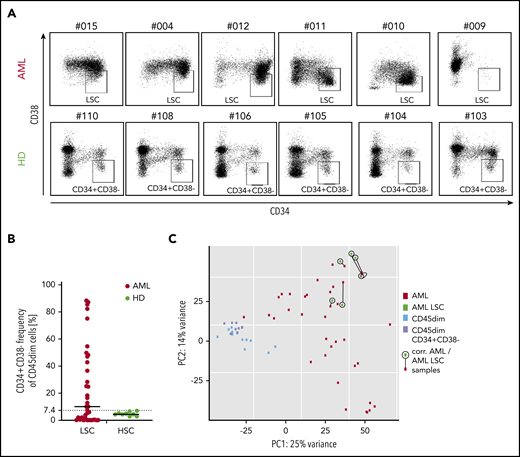
CD34/CD38 LCS-phenotype of pediatric AML samples and pediatric hematopoietic progenitors. (A) While 6 AML CD45dim populations show a very heterogenous CD34 vs CD38 distribution (top panel), their healthy counterparts show a relatively consistent distribution in 6 different cases. (B) Frequency of CD34+CD38− cells in the CD45dim gate in AML patients (median, 10.2%) and healthy donors (median, 4.5%). (C) PCA illustrating 4 populations: bulk AML (AML), AML CD34+CD38− (AML LSC), healthy CD45dim, and healthy CD45dimCD34+CD38−. While both healthy populations cluster together, all 6 AML LSC samples cluster quite close to their corresponding bulk AML sample (as linked by a black line). PC, principal component.
CD34/CD38 LCS-phenotype of pediatric AML samples and pediatric hematopoietic progenitors. (A) While 6 AML CD45dim populations show a very heterogenous CD34 vs CD38 distribution (top panel), their healthy counterparts show a relatively consistent distribution in 6 different cases. (B) Frequency of CD34+CD38− cells in the CD45dim gate in AML patients (median, 10.2%) and healthy donors (median, 4.5%). (C) PCA illustrating 4 populations: bulk AML (AML), AML CD34+CD38− (AML LSC), healthy CD45dim, and healthy CD45dimCD34+CD38−. While both healthy populations cluster together, all 6 AML LSC samples cluster quite close to their corresponding bulk AML sample (as linked by a black line). PC, principal component.
Next, we performed RNA sequencing of the CD34+CD38− population in 6 AML samples and compared them with their parent bulk AML transcriptome. We observed that clustering was primarily determined by patient and not by additional CD34+CD38− sorting (Figure 4C). No significant differential expression was observed between the 6 AML LSC populations and their bulk AML counterparts (not shown). As a result, the majority of AML CD34+CD38− corresponds to the bulk AML populations as analyzed by RNA sequencing.
Combinatorial targeting of CD33/CLEC12A represents the most promising target combination in pediatric AML
The expected efficacy of dual targeting strategies designed to target cells expressing either 1 of 2 selected antigens (OR gated) is determined by the frequency of AML cells that will not express either of the 2 target molecules. In this regard, CD33/CD38 (6% double negatives) and CD33/CLEC12A (8% double negatives) performed best in our flow cytometry analysis (Figure 5A). Next, we analyzed how many AML cells express both target antigens, as dual targeting strategies designed to target cells expressing both selected antigens (AND gated) of AML by CAR T cells require double-positive target cells.9 Interestingly, CD33/CLEC12A (63% double positives) and CD33/CD38 (47% double positives) showed highest coexpression on pediatric AML samples (Figure 5B).

Simultaneous targeting of CD33/CLEC12A yields the highest coverage of pediatric AML, including LSCs. (A) Frequency of AML blasts not expressing either of 2 surface molecules as a measure of AML blasts not covered by combinatorial targeting (mean ± SD). The 7 most promising target combinations are presented. (B) Frequency of AML blasts expressing both targets of 7 promising target combinations (mean ± SD). (C-G) CD45dim cells from 21 samples (not all samples were available for this analysis for technical reasons), with the same cell count per sample, were concatenated into 1 .fcs file, and tSNE parameters were calculated using FlowJo X default settings. Consequently, 5 target combinations (CD33/CD38 in panel C, CD33/CLEC12A in panel D, CD33/EMR2 in panel E, CD33/IL3RA in panel F, and CD33/FLT3 in panel G) are illustrated. Additionally, to depict the coverage of AML LSCs, CD34 and CD38 expression patterns are depicted on the same tSNE map (H).
Simultaneous targeting of CD33/CLEC12A yields the highest coverage of pediatric AML, including LSCs. (A) Frequency of AML blasts not expressing either of 2 surface molecules as a measure of AML blasts not covered by combinatorial targeting (mean ± SD). The 7 most promising target combinations are presented. (B) Frequency of AML blasts expressing both targets of 7 promising target combinations (mean ± SD). (C-G) CD45dim cells from 21 samples (not all samples were available for this analysis for technical reasons), with the same cell count per sample, were concatenated into 1 .fcs file, and tSNE parameters were calculated using FlowJo X default settings. Consequently, 5 target combinations (CD33/CD38 in panel C, CD33/CLEC12A in panel D, CD33/EMR2 in panel E, CD33/IL3RA in panel F, and CD33/FLT3 in panel G) are illustrated. Additionally, to depict the coverage of AML LSCs, CD34 and CD38 expression patterns are depicted on the same tSNE map (H).
With the limitations of LSC gating in mind, we analyzed target expression on CD34+CD38− AML cells using tSNE on concatenated flow cytometry data from AML patients (see “Patients, materials, and methods”). We first plotted the pooled CD34+CD38− AML population (Figure 5H). We used the same tSNE map to demonstrate the expression of the 4 best-performing target combinations (Figure 5C-F; supplemental Figure 1C). While no combination could cover all CD34+CD38− populations, only the addition of CLEC12A and FLT3 could markedly expand CD33 LSC coverage (Figure 5D,G; supplemental Figure 2C-G). In addition, we quantified expression of single target antigens and target combinations on bulk AML (AML CD34+CD38−) and compared it to their healthy counterparts (CD45dim and CD45dimCD34+CD38−). Here, CD33 and CD33/CLEC12A covered fewer cells than on bulk AML but performed best as a single marker and in combination, respectively (supplemental Figure 2A-B).
Finally, 16 out of 21 primary AML samples and 8 out of 15 relapse AML samples contained >50% CD33+CLEC12A+ cells (Figure 6I). Interestingly, CD33/HAVCR2 was useful for cases not amenable to CD33/CLEC12A targeting in AML relapse samples, as 4 AML relapse samples contained >50% CD33+HAVCR2+ cells. However, mean expression of HAVCR2 in CD34+CD38− AML cells was in general lower than CD33 and CLEC12A expression in the same population (supplemental Figure 1B).
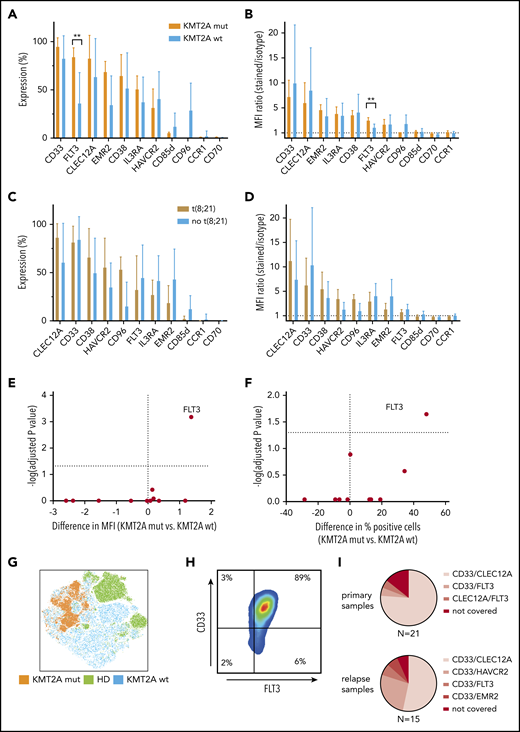
KMT2A-rearranged pediatric AML selectively overexpresses FLT3, and CD33/FLT3 represents a promising immunotherapy combination KMT2A-rearranged AML. (A-B) Eleven surface molecules were quantified by percentage of positive cells (A) and MFI ratio (B) in KMT2A-rearranged AML (n = 5) and KMT2A wild-type AML. The only surface molecule found to be significantly overexpressed in KMT2A-mutated AML was FLT3 (84% vs 36% by percentage, P = .02; 3.4 vs 2.0 by MFI ratio, P = 0,0007). (C-D) Eleven surface molecules were quantified by percentage of positive cells (C) and MFI ratio (D) on t(8;21) and non-t(8;21) AML samples. (E-F) Volcano plots of differential expression in KMT2A-mutated and nonmutated pediatric AML samples. Only FLT3 is significantly overexpressed in KMT2A-mutated AML. (G) tSNE analysis using 36 BM samples, including 9 healthy donors, demonstrates that KMT2A-mutated AML cells cluster within AML samples that themselves cluster from healthy hematopoietic progenitors. (H) When CD45dim cells of 5 KMT2A-mutated AML samples were concatenated and CD33 and FLT3 expression was quantified, an almost homogenous coexpression of CD33 and FLT3 was observed. (I) Setting 50% concomitant expression of 2 target molecules as arbitrary threshold, 16 out of 21 primary AML samples and 8 out of 15 relapse samples could be addressed with CD33/CLEC12A targeting. CD33/HAVCR2 targeting was particularly effective in AML relapse samples that could not be addressed with CD33/CLEC12A targeting, as 4 out of 7 samples were >50% double positive. wt, wild-type.
KMT2A-rearranged pediatric AML selectively overexpresses FLT3, and CD33/FLT3 represents a promising immunotherapy combination KMT2A-rearranged AML. (A-B) Eleven surface molecules were quantified by percentage of positive cells (A) and MFI ratio (B) in KMT2A-rearranged AML (n = 5) and KMT2A wild-type AML. The only surface molecule found to be significantly overexpressed in KMT2A-mutated AML was FLT3 (84% vs 36% by percentage, P = .02; 3.4 vs 2.0 by MFI ratio, P = 0,0007). (C-D) Eleven surface molecules were quantified by percentage of positive cells (C) and MFI ratio (D) on t(8;21) and non-t(8;21) AML samples. (E-F) Volcano plots of differential expression in KMT2A-mutated and nonmutated pediatric AML samples. Only FLT3 is significantly overexpressed in KMT2A-mutated AML. (G) tSNE analysis using 36 BM samples, including 9 healthy donors, demonstrates that KMT2A-mutated AML cells cluster within AML samples that themselves cluster from healthy hematopoietic progenitors. (H) When CD45dim cells of 5 KMT2A-mutated AML samples were concatenated and CD33 and FLT3 expression was quantified, an almost homogenous coexpression of CD33 and FLT3 was observed. (I) Setting 50% concomitant expression of 2 target molecules as arbitrary threshold, 16 out of 21 primary AML samples and 8 out of 15 relapse samples could be addressed with CD33/CLEC12A targeting. CD33/HAVCR2 targeting was particularly effective in AML relapse samples that could not be addressed with CD33/CLEC12A targeting, as 4 out of 7 samples were >50% double positive. wt, wild-type.
KMT2A-mutated AML samples can additionally be addressed with CD33/FLT3 targeting
As we had observed clustering of KMT2A-mutated and t(8;21) AML samples by PCA of RNA-sequencing data, we investigated whether immunotargets showed differential surface protein expression in those genetic AML groups. While no altered surface expression profile could be observed in t(8;21) and AML samples without t(8;21), KMT2A-mutated AML samples displayed significantly higher FLT3 surface expression compared with KMT2A wild-type AML (Figure 6A-D). FLT3 surface overexpression in KMT2A-rearranged AML was significantly higher, as measured by both the MFI ratio and percentage of positive cells (Figure 6E-F). Additionally, all 5 KMT2A mutated AML samles were homogenously positive for CD33 (Figure 6H). Moreover, KMT2A-mutated samples cluster apart in a tSNE analysis of samples from both KMT2A wild-type AML patients and healthy donors, thereby supporting the opportunity of a specific immunotherapy for KMT2A-mutated AML (Figure 6G).
Discussion
Standard treatment of AML is based on high-dose chemotherapy regimens using agents such as cytarabine and anthracyclines. Targeted T-cell–based immunotherapies offer great promise for AML treatment but still face challenges in clinical application.33 Bispecific T-cell engagers and antibodies are in clinical development for use as off-the-shelf products. CAR T cells combine specificity of antibodies with strong T-cell effector function. However, challenges related to heterogeneous target expression by AML, on-target off-leukemia side effects, and CAR T-cell persistence remain to be solved. Recent insights into AML biology have documented a striking heterogeneity of AML,34 and the diversity of pediatric AML has been recently demonstrated in a large study.20 Consequently, it is questionable whether concepts for synthetic immunotherapy of AML can be easily translated from adult to pediatric patients. In the current study, we investigated which surface molecules are suitable as targets for synthetic immunotherapy in pediatric AML. Targets for adult AML synthetic immunotherapy have been studied extensively in recent years,9,10 but only limited data are available on immunotargets of pediatric AML. Several clinical trials using CAR T cells as means of synthetic immunotherapy are currently evaluating the feasibility of AML targeting using single targets like CD33 and IL3RA.33
We show that pediatric AML transcriptomes cluster separately from healthy pediatric hematopoietic progenitors by PCA. KMT2A-mutated infant AML samples form a separate cluster within pediatric AML samples. When we analyzed a set of 35 target molecules (supplemental Table 1), recently reported as most promising AML targets in adults, we observed expression patterns in pediatric AML that are different from data in adult AML. In our pediatric AML cohort, IL3RA (CD123) is not overexpressed on the RNA level compared with healthy hematopoietic progenitors, and cell surface expression on pediatric AML blasts was limited to a minority of AML samples. Therefore, IL3RA does not appear to be a suitable target for pediatric AML. Additional molecules like CCR1 and LILRB2, which have been described as promising targets in adult AML, were not found to be enriched in the transcriptome of our pediatric cohort or expressed on the surface of AML blasts. Other reported genes, such as FOLR2 and LILRA6, did not show enrichment in pediatric AML when compared with healthy hematopoietic progenitors. Genes such as ITGB5 and CD38 showed high expression levels in AML but are also expressed in healthy nonhematopoietic tissues and are consequently not attractive as AML immunotargets.
Our data show a high coexpression of CD33 and CLEC12A in pediatric AML blasts. Both molecules are highly enriched in pediatric AML by RNA sequencing, overexpressed on AML blasts on protein level, and show only low expression in healthy nonhematopoietic tissues. Comparing different combinations of overexpressed molecules, the combination of CLEC12A and CD33 has the best coverage and specificity. Therefore, CLEC12A and CD33 appear to be the preferred targets in pediatric AML for combinatorial approaches.
The concept of LSCs derived from serial transfer experiments and targeting LSCs has been discussed as a requirement for AML immunotargeting.10 We observed that defining CD34+CD38− LSCs has its limitations in BM of pediatric AML patients, as phenotypic discrimination of LSCs remained questionable in the majority of patients. Therefore, CD45dim gating remain the best suitable gating for identification of potential target molecules in a comparison of AML with healthy counterparts. In contrast, a multiparameter gating of 5 or 6 surface markers such as CD45dimCD34+CD38−CD45RA−CD90+ for HSCs can provide the most specific information about potential on-target off-leukemia effects on healthy hematopoietic precursor subpopulations. However, even with these limitations of phenotypic LSC gating, CLEC12A and CD33 showed best coverage in the CD34+CD38− AML subpopulation.
Synthetic immunotherapy has been defined as a treatment that leads to an immune response that was not present prior to an intervention in an individual patient.35 This concept is particularly attractive for pediatric AML as a malignancy with particularly low mutational load.36 Recent reports on clinical efficacy have underlined that CAR T cells are the most potent effectors of synthetic immunotherapy. For development of CAR T-cell therapies against pediatric AML, our data provide information on coverage using target expression profiles on AML and information on potential safety in terms of expression on healthy hematopoietic cell populations. CD33 has been targeted for many years using monoclonal antibodies against CD33 coupled to a cytotoxic agent.37 Moreover, transient efficacy of CD33-directed CAR T cells was documented in a 41-year-old male.38 We confirm CD33 expression on hematopoietic precursors in BM of healthy children from early HSCs over MPPs, MLPs, CMPs, and GMPs up to monocytes. In line with this finding, the main side effect of CD33-directed immunotherapy is pancytopenia, and hence, current CD33-targeted CAR approaches rely on subsequent stem cell transplantation.12,39-41 In our pediatric healthy BM cohort, CLEC12A expression is absent on early hematopoietic precursors such as HSCs, and increases in more differentiated myeloid subpopulations such as MLPs, CMPs, GMPs, and monocytes. CLEC12A-directed CAR T-cell therapy has been recently reported to induce remission in a pediatric patient with secondary AML, who was in remission at the time of publication.42 Additionally, there is a clinical study employing CD33-CLEC12A compound CAR T cells for AML therapy with subsequent stem cell transplant (NCT03795779). So far, 2 patients with successful treatment including subsequent allogeneic stem cell transplant have been reported, a 6-year old girl and a 44-year old man.43
Dual targeting in cancer immunotherapy can be designed to target cells expressing both antigens (AND gated) or either 1 of 2 selected antigens (OR gated).44-46 The first approach will increase specificity for malignant cells at the expense of decreased AML coverage. The latter approach will increase the coverage percentage of targeted malignant cells at the expense of increased off-leukemia effects. In our analysis, the combination of CD33 and CLEC12A would cover >90% of AML blasts across 36 samples, analyzed in an OR-gated combinatorial setup, addressing both single-positive (CD33 or CLEC12A) and double-positive cells. Application of an AND-gated CAR, such as the compound CAR45 approach, would result in coverage of 22 out of 36 patients, assuming that the expression of CD33 and CLEC12A on 75% of AML cells is sufficient for AML eradication. Based on our data presented here, one promising approach for pediatric AML CARs would be an AND-gated CD33-CLEC12A CAR. In vivo data and clinical data will have to clarify the requirement of a safety switch or subsequent stem cell transplantation. Second, an OR-gated CD33-CLEC12A CAR will definitively require a safety switch for CAR depletion and/or subsequent stem cell transplantation (Figure 3E).
The AML transcriptome showed KMT2A-status–specific clustering as analyzed with PCA. Importantly, we show significantly increased FLT3 expression on KMT2A-rearranged samples, which has also been recently reported in a study from the Children’s Oncology Group.47 In fact, all KMT2A-rearranged AML samples studied were homogenously high positive for both FLT3 and CD33, indicating that dual targeting of CD33 and FLT3 might be a promising approach to KMT2A-rearranged infant AML immunotherapy. Current reports suggest that FLT3 genotype does not influence FLT3 surface expression.47,48 However, as CD33 and FLT3 are both expressed on HSCs, this approach can only be envisioned in a concept including CAR depletion via a safety switch and subsequent allogeneic stem cell transplantation (Figure 3F).
In summary, combinatorial targeting of pediatric AML with immunotherapy will require an age-specific approach for children and adolescents and a genotype-specific selection of target antigens for subgroups. Clinical efficacy of combinatorial immunotherapy against pediatric AML will have to be confirmed in clinical trials.
For original data, please contact the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients and their families for participating in the study and Nadine Stoll for excellent technical assistance.
This work was supported by the Kinderkrebshilfe Ebersberg eV, Bettina Braeu Stiftung, Dr Sepp und Hanne Sturm Gedaechtnisstiftung, Gertrud und Hugo Adler Stiftung, Renate & Roland Gruber Stiftung and Otto-Hellmeier Stiftung. S.W. was supported by the Else-Kröner-Fresenius Stiftung, P.R. by the Kind-Philipp-Stiftung, J.W. by Deutsche José Carreras Leukämie-Stiftung. and D.S. by the German Cancer Research Center/German Cancer Consortium.
Authorship
Contribution: S.W. and T.F. set up the concept of this study; design and approach of experiments was done by S.W., F.B., and T.F.; RNA sequencing was done by S.W., M.R., and C.K.; V.B., M.H.A., and I.S. provided patient data, BM, and healthy donor samples; P.R., J.W., and S.W. processed patient samples; M.H. performed bioinformatics analysis of sequencing data; S.W., M.H., and P.R. analyzed the data; S.W. and T.F. prepared the manuscript; and all authors reviewed and approved the final manuscript.
Conflict-of-interest disclosure: M.H.A. declares Amgen stock ownership. The remaining authors declare no competing financial interests.
Correspondence: Tobias Feuchtinger, Department of Pediatric Hematology, Oncology, Hemostaseology and Stem Cell Transplantation, Dr von Hauner Children’s Hospital, Ludwig Maximilian University Munich, Lindwurmstrasse 4, 80337 Munich, Germany; e-mail: tobias.feuchtinger@med.uni-muenchen.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal