

Key Points
Treatment of RS PDX models with Duv and Ven combination induces tumor regression and prolongs survival.
Synergism between Duv and Ven is mediated by GSK3β, a key player at the crossroad between PI3K and apoptotic pathways.

Abstract
A small subset of cases of chronic lymphocytic leukemia undergoes transformation to diffuse large B-cell lymphoma, Richter syndrome (RS), which is associated with a poor prognosis. Conventional chemotherapy results in limited responses, underlining the need for novel therapeutic strategies. Here, we investigate the ex vivo and in vivo efficacy of the dual phosphatidylinositol 3-kinase-δ/γ (PI3K-δ/γ) inhibitor duvelisib (Duv) and the Bcl-2 inhibitor venetoclax (Ven) using 4 different RS patient-derived xenograft (PDX) models. Ex vivo exposure of RS cells to Duv, Ven, or their combination results in variable apoptotic responses, in line with the expression levels of target proteins. Although RS1316, IP867/17, and RS9737 cells express PI3K-δ, PI3K-γ, and Bcl-2 and respond to the drugs, RS1050 cells, expressing very low levels of PI3K-γ and lacking Bcl-2, are fully resistant. Moreover, the combination of these drugs is more effective than each agent alone. When tested in vivo, RS1316 and IP867/17 show the best tumor growth inhibition responses, with the Duv/Ven combination leading to complete remission at the end of treatment. The synergistic effect of Duv and Ven relies on the crosstalk between PI3K and apoptotic pathways occurring at the GSK3β level. Indeed, inhibition of PI3K signaling by Duv results in GSK3β activation, leading to ubiquitination and subsequent degradation of both c-Myc and Mcl-1, making RS cells more sensitive to Bcl-2 inhibition by Ven. This work provides, for the first time, a proof of concept of the efficacy of dual targeting of PI3K-δ/γ and Bcl-2 in RS and providing an opening for a Duv/Ven combination for these patients. Clinical studies in aggressive lymphomas, including RS, are under way. This trial was registered at www.clinicaltrials.gov as #NCT03892044.
Introduction
Richter syndrome (RS), the transformation of chronic lymphocytic leukemia (CLL) into an aggressive lymphoma, occurs ∼2% to 10% of patients with CLL.1 In the majority of cases, CLL evolves into a diffuse large B-cell lymphoma (DLBCL) that maintains a clonal relationship with the original leukemic phase, whereas the rest of the patients develop a Hodgkin lymphoma variant.2 Survival of patients with RS is generally poor, and subjects carrying selective chromosomal aberrations or being clonally related to CLL experience the worst prognosis and outcome.3,4
Several genetic and immune factors may contribute to the transformation. In recent years, additional risk factors have been identified, such as TP53 disruption, NOTCH1 mutation, CDKN2A loss, and MYC activation.2,5-7 In addition, biased usage of subset 8 V4-39 stereotyped immunoglobulin gene increases the risk of RS development by 24-fold,8,9 suggesting a driving role of B-cell receptor (BCR) signaling in transformation. Overall, the molecular profile of RS is heterogeneous, lacking a unifying lesion, and does not overlap with the genetics of de novo DLBCL.6 Deregulation of the underlying transcriptional programs and signaling pathways may account for the aggressive clinical phenotype of RS.3
Conventional chemotherapy appears to have only partial and transitory effects, with overall response rates of ∼40%,2,7 prompting the need for novel therapeutic strategies that target the main elements that drive RS transformation and biology. Despite improvements in CLL treatment, mainly based on targeted therapies, results of these novel agents in RS are still controversial, with mostly anecdotic experiences.3
Indeed, therapy of patients with CLL has radically changed since the approval of the BTK inhibitor, ibrutinib, in February 2012, with a new wave of small molecules replacing chemo-immunotherapy approaches. These same drugs are being explored for patients with RS. However, so far, they are associated with poor responses, suggesting that there may be intrinsic resistance to targeted drugs in patients with RS who have been heavily pretreated during the CLL phase. So far, the majority of clinical data show only limited responses to the BTK inhibitors ibrutinib10-12 and acalabrutinib in patients with RS.13 No data are yet available on the efficacy of the Bcl-2 inhibitor, venetoclax (Ven), in RS, which is currently in a phase 2 clinical trial in combination with chemotherapy (NCT03054896).14 The use of the phosphatidylinositol 3-kinase-δ (PI3K-δ) inhibitor, idelalisib, alone or in combination with ibrutinib, has been proposed for overcoming ibrutinib resistance because in vitro inhibition of PI3K-δ has been shown to reverse ibrutinib resistance in mantle cell lymphoma, but further investigations are needed.15,16
A better understanding of the biology of the disease together with an in-depth molecular profiling of RS cells to define the relevant survival pathways is a necessary step to highlight additional opportunities for a rational development of combination therapies.
The need for novel and additional therapeutic combinations to treat patients with RS and preliminary data from a phase 1 clinical trial on patients with relapsed/refractory CLL/small lymphocytic leukemia,17 prompted us to evaluate the expression of PI3K-δ/γ and Bcl-2 molecules in primary RS samples and RS patient-derived xenograft (RS-PDX) models. We therefore examined the ex vivo and in vivo efficacy of duvelisib (Duv), a dual PI3K-δ/γ inhibitor, and Ven, a Bcl-2 inhibitor, alone or in combination, as a potential therapeutic approach for RS.
Explanation of novelty
Our findings indicate that the combined use of highly selective inhibitors targeting PI3K-γ/δ and Bcl-2 is highly effective in RS xenograft models, provided the lymphoma expresses both target molecules. They also argue in favor of enrollment of patients with RS, characterized by PI3K and Bcl-2 expression, in the ongoing clinical trial investigating the efficacy of these drugs.18
Methods
RS-PDX models
RS-PDX models were established and maintained as previously described.19 Briefly, for the subcutaneous (SC) model, 5 × 106 RS cells were resuspended in Matrigel/culture media (1:1 ratio; Corning, Milan, IT) and injected double flank in 8-week-old nonobese diabetic/severe combined immunodeficiency/γ chain−/− (NSG) immunocompromised mice. For the IV model, 10 × 106 RS cells were resuspended in phosphate-buffered saline and injected in the tail vein of NSG mice. The Institutional Animal Care and Use Committee approved all the experiments involving mice. Mice were treated following the European guidelines and with the approval of the Italian Ministry of Health (authorization #664/2020-PR).
Ex vivo treatment of RS cells
RS cells, freshly purified as a single-cell suspension from tumor masses, were exposed to drugs for the indicated time points, depending on the read-out considered. Specifically, we used: Duv (5 μM; 3, 6, 24, 48 hours); Ven (25 nM; 3, 6, 24, 48 hours); Acalisib (5 μM; 6 hours); IPI-549 (5 μM; 6 hours); MG-132, a proteasome inhibitor (10 μM; 3-6 hours), and tideglusib, a GSK3β inhibitor (2.5 μM; 3-6 hours). Duv was refilled every 24 hours of culture. After drug treatment, cells were processed as indicated in the following section.
In vivo treatments
For the SC experiments, RS cells were left to engraft and when tumor masses were ∼0.2 cm3, animals were randomly assigned to 4 different groups and treated by oral gavage for 10 consecutive days with vehicle (5% dimethyl sulfoxide, 90% PEG300, and 5% Tween 80; all from Sigma-Aldrich), Duv (100 mg/kg), Ven (50 mg/kg), or their combination. During drug administration and after treatment discontinuation, mice were monitored and tumor masses regularly measured twice a week. Mice were euthanized when tumor masses were ∼1 cm3 or 60 days postinjection. In another experimental setting (short-term study), when tumor masses were ∼0.1 cm3 mice were treated for 5 consecutive days and at the end of the treatment, euthanized and compared for tumor growth, cell viability, and apoptosis.
For the IV experiments, RS cells were injected, left to engraft for 10 to 14 days, and then animals were randomly assigned to 4 different groups and treated as indicated for the SC experiments. During drug administration and after treatment discontinuation, mice were monitored and weighed twice a week. Mice were euthanized when exhibiting clear signs of disease, including cachexia and body weight loss ∼20%.
Immunoprecipitation
For immunoprecipitation experiments, RS1316 cells were treated as indicated previously. Cells were then harvested and lysed. A total of 500 µg of each lysate was precleared with immunoglobulin G–coated protein A magnetic sepharose beads (30 minutes, 4°C). Then, precleared lysates were incubated with anti-Mcl-1– and anti-c-Myc–coated beads (3 hours, 4°C). Immunocomplexes were then washed 3 consecutive times with a Tris buffer (50 mM Tris, 150 mM NaCl, pH 7.5), eluted with sodium dodecyl sulfate 2%, and analyzed by western blot.
Additional details on materials and methods are available as supplemental Data on the Blood Web site.
Results
PI3K subunits and antiapoptotic proteins are expressed at variable levels in primary cells and in RS-PDX models
Investigating drug repositioning for patients with RS, we evaluated PI3K-δ/γ and Bcl-2 expression by immunohistochemistry in a cohort of 24 RS primary samples showing a clear cytoplasmic positivity for PI3K-δ/γ and Bcl-2 in the majority of cases (Figure 1A). Specifically, all the samples analyzed were invariably positive for PI3K-δ, whereas PI3K-γ and Bcl-2 were expressed in 18 of 24 (75%) samples (Figure 1B). These data suggested that PI3K and Bcl-2 targeting was worthy to be explored because it may be useful for a significant subset of patients with RS.
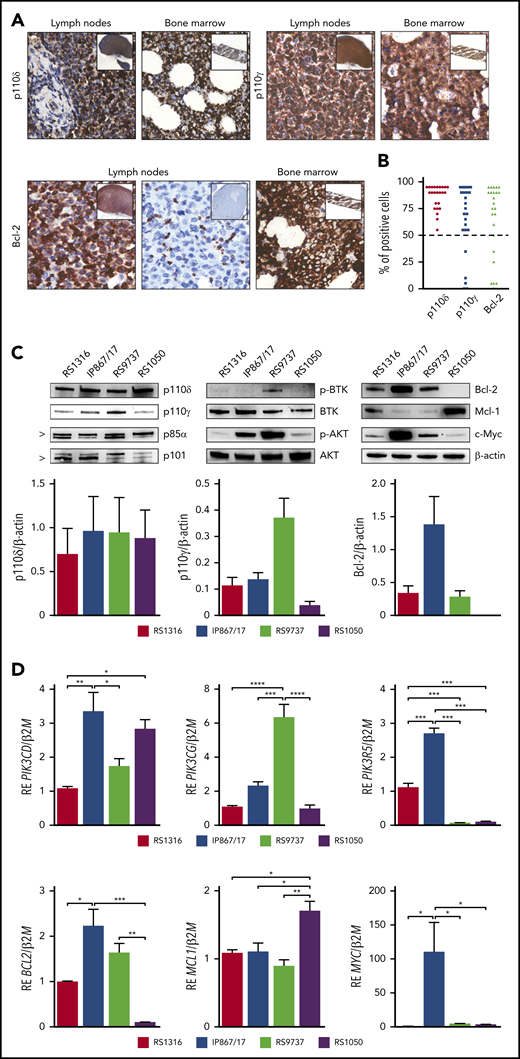
RS-PDX models molecular profiling. (A) Representative immunohistochemistry images of PI3K-δ/γ and Bcl-2 expression in lymph nodes and bone marrow biopsies of patients with RS (magnification ×40; insets ×4). (B) Percentage of PI3K-δ/γ and Bcl-2 positive cells in a cohort of 24 RS primary samples. Dashed line indicates the 50% threshold used to define positive samples. (C) Western blot panels showing expression of proteins belonging to the BCR and PI3K cascades (p110δ, p110γ, p85α, p101, p-BTK, BTK, p-AKT, AKT, c-Myc) and apoptotic pathway (Bcl-2, Mcl-1) in the available RS-PDX models (RS1316, IP867/17, RS9737, RS1050). Bar plots represent intensity of p110δ, p110γ, and Bcl-2 bands in 4 independent experiments. Band intensities were measured using Image Lab and normalized on β-actin. Data are reported as mean ± standard error of the mean (SEM). (D) Quantitative reverse transcriptase polymerase chain reaction analyses of members of the PI3K family (PIK3CD, PIK3CG, PIK3R5), apoptotic pathway (BCL2, MCL1), and MYC. Messenger RNA expression is normalized over β-2-microglobulin (B2M). Data are reported as mean ± SEM. RE, relative expression. Statistical analysis was performed using 1-way analysis of variance (ANOVA); *P < .05, **P < .01, ***P < .001, ****P < .0001.
RS-PDX models molecular profiling. (A) Representative immunohistochemistry images of PI3K-δ/γ and Bcl-2 expression in lymph nodes and bone marrow biopsies of patients with RS (magnification ×40; insets ×4). (B) Percentage of PI3K-δ/γ and Bcl-2 positive cells in a cohort of 24 RS primary samples. Dashed line indicates the 50% threshold used to define positive samples. (C) Western blot panels showing expression of proteins belonging to the BCR and PI3K cascades (p110δ, p110γ, p85α, p101, p-BTK, BTK, p-AKT, AKT, c-Myc) and apoptotic pathway (Bcl-2, Mcl-1) in the available RS-PDX models (RS1316, IP867/17, RS9737, RS1050). Bar plots represent intensity of p110δ, p110γ, and Bcl-2 bands in 4 independent experiments. Band intensities were measured using Image Lab and normalized on β-actin. Data are reported as mean ± standard error of the mean (SEM). (D) Quantitative reverse transcriptase polymerase chain reaction analyses of members of the PI3K family (PIK3CD, PIK3CG, PIK3R5), apoptotic pathway (BCL2, MCL1), and MYC. Messenger RNA expression is normalized over β-2-microglobulin (B2M). Data are reported as mean ± SEM. RE, relative expression. Statistical analysis was performed using 1-way analysis of variance (ANOVA); *P < .05, **P < .01, ***P < .001, ****P < .0001.
We then moved to examine 4 RS-PDXs characterized by different mutational and molecular profiles that fully recapitulate the heterogeneity of the disease.19,20 Expression of PI3K catalytic (p110δ and p110γ) and regulatory (p85α and p101) subunits, phosphorylated (p-) and total BTK and AKT, the apoptotic pathway proteins Bcl-2 and Mcl-1, and c-Myc was evaluated by western blot. The p110δ and p85α subunits were homogenously expressed at high levels by all RS-PDX models, whereas the catalytic p110γ and the regulatory p101 subunits displayed heterogeneous levels, with RS1050 showing the lowest expression (Figure 1C). Quantitative reverse transcriptase polymerase chain reaction analysis confirmed these results (Figure 1D). This variable expression correlated with a different activation status of BTK and AKT, an upstream and a downstream player of the PI3K pathway, respectively, which are mainly active in the IP867/17 and RS9737 models. We also checked the expression of c-Myc, a known downstream element of the BCR and PI3K pathway and a transcription factor that regulates the expression of proliferative and survival genes, among others.21,22 c-Myc was overexpressed in the IP867/17 model both at the protein and messenger RNA levels, whereas it was present at lower levels in the other models (Figure 1C-D).
When looking at the apoptotic pathway, RS1316, IP867/17, and RS9737 expressed high levels of the antiapoptotic protein Bcl-2, which was completely absent in RS1050, characterized, on the contrary, by the expression of Mcl-1, as confirmed by reverse transcriptase polymerase chain reaction results (Figure 1C-D).
These results indicate that our RS-PDXs recapitulate patients expression patterns and can therefore be used further for in vitro and in vivo testing of PI3K and Bcl-2 targeting.
Duv and Ven synergistically induce apoptosis in PI3K and Bcl-2 expressing RS cells ex vivo
Given the expression of PI3K and Bcl-2, we first evaluated the effect of their inhibition in an ex vivo setting, using Duv and Ven, alone and in combination. Flow cytometry analysis of apoptosis indicated that RS cells were differently sensitive to these drugs. Indeed, RS1316 was the best responder to both single agents and their combination, starting 24 hours after treatment and peaking at 48 hours. IP867/17 and RS9737 showed a higher sensitivity to Ven compared with Duv, at least at 24 hours, with the combination resulting in a significant induction of apoptosis that was more pronounced at 48 hours of exposure. On the contrary, and in line with the lack of PI3K and Bcl-2 expression, RS1050 was completely insensitive to both drugs (Figure 2A; supplemental Figure 1A). These results were corroborated at the protein level, highlighting Caspase-3 and PARP1 cleavage at both time points (Figure 2B; supplemental Figure 1B).
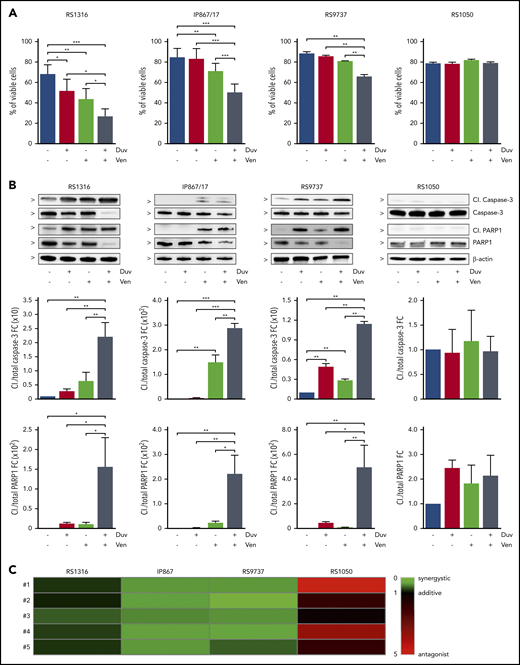
Induction of apoptosis in RS cells ex vivo treated for 24 hours with Duv, Ven, and their combination. (A) Percentage of viable RS cells after 24 hours’ treatment with vehicle (blue), Duv (red), Ven (green), and their combination (gray). Data are reported as mean ± SEM. (B) Expression levels of cleaved (Cl.) and total Caspase-3 and PARP1 in RS cells after 24 hours’ treatment with Duv, Ven, and their combination. Bar plots represent intensity of proteins bands in 5 independent experiments. Band intensities were measured using Image Lab, normalized on β-actin, and plotted as fold change (FC) over the band of vehicle treated cells. Data are reported as mean ± SEM. (C) CI analysis. CI was calculated using the Bliss Independence model to compute the effect of Duv and Ven combination. Synergistic effect is defined as CI < 1 (green), while additive effect is CI around 1 (black) and antagonist effect is CI > 1 (red). Statistical analysis was performed using 1-way ANOVA; *P < .05, **P < .01, ***P < .001, ****P < .0001.
Induction of apoptosis in RS cells ex vivo treated for 24 hours with Duv, Ven, and their combination. (A) Percentage of viable RS cells after 24 hours’ treatment with vehicle (blue), Duv (red), Ven (green), and their combination (gray). Data are reported as mean ± SEM. (B) Expression levels of cleaved (Cl.) and total Caspase-3 and PARP1 in RS cells after 24 hours’ treatment with Duv, Ven, and their combination. Bar plots represent intensity of proteins bands in 5 independent experiments. Band intensities were measured using Image Lab, normalized on β-actin, and plotted as fold change (FC) over the band of vehicle treated cells. Data are reported as mean ± SEM. (C) CI analysis. CI was calculated using the Bliss Independence model to compute the effect of Duv and Ven combination. Synergistic effect is defined as CI < 1 (green), while additive effect is CI around 1 (black) and antagonist effect is CI > 1 (red). Statistical analysis was performed using 1-way ANOVA; *P < .05, **P < .01, ***P < .001, ****P < .0001.
To better understand whether Duv and Ven had a synergistic or additive effect on RS cells, their combination index (CI) was calculated. Data indicated that induction of apoptosis by the 2 drugs used in combination was synergistic, with a CI < 1 in RS1316 (CI = 0.794), IP867/17 (CI = 0.426), and RS9737 (CI = 0.404). In contrast, and as expected, the CI in RS1050 was 2.718, confirming that these cells did not show significant responses to these drugs (Figure 2C).
Because there are other PI3K inhibitors on the market that can selectively block the γ or δ subunits, we also tested their ex vivo effects on RS cells, either alone or in combination with Ven. Data indicate that these drugs induced similar responses to those obtained with Duv or Duv/Ven (supplemental Figure 2), suggesting that both subunits are active in this context. Given the overlapping results, we decided to further concentrate on Duv to inhibit activation of PI3K coming from different molecular pathways.
Taken together, these results indicate that the molecular profile of RS cells can be used to predict sensitivity to Duv and Ven, which act synergistically when PI3K-δ/γ and Bcl-2 are expressed.
Duv and Ven combination induces apoptosis of RS cells in vivo
After evidence of ex vivo induction of apoptosis on RS cells following drugs exposure, we investigated their ability to induce similar effects in a pilot short-term in vivo experiment, where all mice were euthanized at the same time point and we specifically evaluated the activation of Caspase-3. To this purpose, NSG mice were SC injected with RS1316 cells, the best ex vivo responder model. When tumor masses became palpable (∼0.2 cm3), animals were randomized and treated for 5 consecutive days by oral gavage with vehicle, Duv (100 mg/kg), Ven (50 mg/kg), or the combination of Duv/Ven (Figure 3A). In line with ex vivo data, Duv- and Ven-treated mice showed a significant reduction of tumor volume when compared with the vehicle group. However, the combination of the 2 drugs turned out to be the most effective treatment with 2 of 3 mice being disease-free and the remaining one presenting with very small tumor masses compared with vehicle or single agent-treated animals (0.16 vs 0.39 cm3, respectively; P = .0066; Figure 3B). Hematoxylin/eosin staining of the tumor masses indicated that treatment with drugs alone or in combination reduced the number of RS cells with a concomitant increase of necrotic and fibrotic tissue. Moreover, immunohistochemistry data showed positive staining for cleaved Caspase-3, which was comparable between Duv and Ven as single agents, but higher in the combination, confirming a synergistic effect when blocking both targets (CI = 0.78; Figure 3C-D).
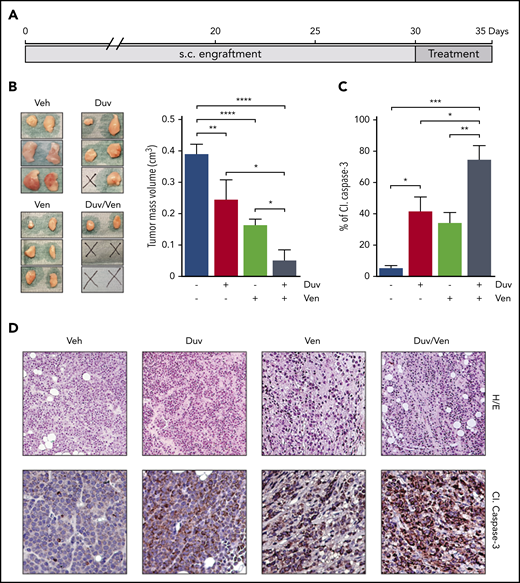
Tumor growth inhibition and induction of apoptosis in RS1316 SC model treated with Duv, Ven and their combination. (A) Experimental design: RS1316 cells were SC injected and left to engraft. Mice were randomized in 4 treatment groups, treated for 5 consecutive days, and euthanized at the same time point. (B) Pictures of tumor masses (left) and summary graph of their volume (right) at the end of the experiment. Data are reported as mean ± SEM. (C) Quantification of cleaved (Cl.) Caspase-3 signal, obtained by immunohistochemistry performed on tumor masses, represented as percentage of positive area. Data are reported as mean ± SEM. (D) Hematoxylin/eosin (H/E, ×20) and cleaved (Cl.) Caspase-3 IHC staining (magnification ×40). Veh: vehicle. Statistical analysis was performed using 1-way ANOVA; *P < .05, **P < .01, ***P < .001, ****P < .0001.
Tumor growth inhibition and induction of apoptosis in RS1316 SC model treated with Duv, Ven and their combination. (A) Experimental design: RS1316 cells were SC injected and left to engraft. Mice were randomized in 4 treatment groups, treated for 5 consecutive days, and euthanized at the same time point. (B) Pictures of tumor masses (left) and summary graph of their volume (right) at the end of the experiment. Data are reported as mean ± SEM. (C) Quantification of cleaved (Cl.) Caspase-3 signal, obtained by immunohistochemistry performed on tumor masses, represented as percentage of positive area. Data are reported as mean ± SEM. (D) Hematoxylin/eosin (H/E, ×20) and cleaved (Cl.) Caspase-3 IHC staining (magnification ×40). Veh: vehicle. Statistical analysis was performed using 1-way ANOVA; *P < .05, **P < .01, ***P < .001, ****P < .0001.
In vivo combination of Duv and Ven significantly blocks tumor growth in SC RS-PDX models
We then expanded our preliminary data on in vivo efficacy of these drugs in all the available RS-PDXs in a long-term experiment, exploiting the SC model. In this setting, treatment started when masses were ∼0.2 cm3 and continued for 10 consecutive administrations, with the same doses as indicated previously (Figure 4). Mice were monitored for tumor growth kinetics by measuring tumor mass volume twice a week.
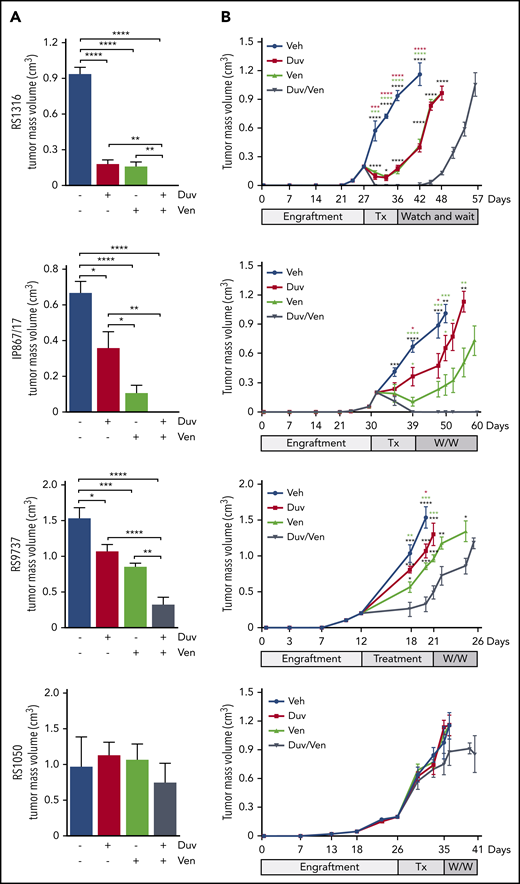
Tumor growth kinetics of RS-PDX SC models treated with Duv, Ven, and their combination. (A) Tumor volume at the end of treatments in the 4 RS-PDX SC models. Data are reported as mean ± SEM. (B) Tumor growth kinetic of RS-PDX SC models treated with vehicle (Veh; blue), Duv (red), Ven (green), and their combination (Duv/Ven; gray). Data are reported as mean ± SEM. Tx, treatment; W/W, watch and wait. Statistical analysis was performed using 1-way ANOVA (A) and a multiple Student t test (B); *P < .05, **P < .01, ***P < .001, ****P < .0001.
Tumor growth kinetics of RS-PDX SC models treated with Duv, Ven, and their combination. (A) Tumor volume at the end of treatments in the 4 RS-PDX SC models. Data are reported as mean ± SEM. (B) Tumor growth kinetic of RS-PDX SC models treated with vehicle (Veh; blue), Duv (red), Ven (green), and their combination (Duv/Ven; gray). Data are reported as mean ± SEM. Tx, treatment; W/W, watch and wait. Statistical analysis was performed using 1-way ANOVA (A) and a multiple Student t test (B); *P < .05, **P < .01, ***P < .001, ****P < .0001.
In the RS1316 model, at the end of drug administration, Duv- and Ven-treated mice showed a significant reduction in tumor volume (0.181 ± 0.086 cm3 and 0.160 ± 0.110 cm3, respectively; P < .0001) compared with the vehicle-treated group (0.937 ± 0.132 cm3). In contrast, no tumor masses could be detected in mice treated with the Duv/Ven combination (Figure 4A). Mice were then monitored for tumor regrowth and euthanized when masses reached ∼1 cm3. Tumor masses resumed to grow right after the end of treatment in Duv- and Ven-treated mice, with the same kinetic of the vehicle-treated group, whereas the Duv/Ven-treated mice showed a delay of 9 days and were euthanized 15 days after vehicle-treated mice (Figure 4B).
Mice injected with the IP867/17 model showed significant responses to both agents (0.360 ± 0.258 cm3 in Duv-treated mice and 0.104 ± 0.127 cm3 in Ven-treated group; P < .022 and P < .0001, respectively) compared with vehicle-treated mice (0.669 ± 0.148 cm3), with no evidence of tumor masses in Duv/Ven combination group (Figure 4A). Moreover, all the Duv/Ven-treated mice remained disease-free until the end of experiment, 3 weeks after drugs discontinuation, whereas tumor masses resumed growing right after the end of treatment in Duv- and Ven-treated mice (Figure 4B).
Using the RS9737, the fastest growing model, we observed partial responses in the Duv and Ven groups compared with vehicle-treated mice (Duv: 1.071 ± 0.269 cm3, P = .018; Ven: 0.859 ± 0.127 cm3, P = .0003; vehicle: 1.537 ± 0.335 cm3), with a marked tumor reduction obtained in the Duv/Ven combination group (0.328 ± 0.290 cm3, P < .0001; Figure 4A). Treatment interruption in these animals was followed by rapid tumor regrowth, with only 6 days’ delay for the combination group compared with vehicle-treated ones (Figure 4B).
It is worth underlining that, in all these models, the combination of Duv and Ven resulted in more pronounced effects compared with each single drug, reaching statistical significance.
Finally, we tested the in vivo efficacy of Duv and Ven in RS1050, characterized by very low levels of PI3K-γ and lacking Bcl-2. As expected, and in line with ex vivo data, all treated mice failed to respond to the drugs (Duv/Ven: 0.971 ± 0.413; Duv: 1.131 ± 0.180 cm3; Ven: 1.068 ± 0.214 cm3; vehicle: 0.971 ± 0.168 cm3; Figure 4A). Moreover, because of the overlapping tumor growth kinetics, no significant delay among treated groups was observed after treatment interruption (Figure 4B).
In vivo treatment does not result in drug resistance
To exclude that tumor regrowth was due to drug-resistance phenomena or clonal selection of drug-insensitive neoplastic cells during in vivo drug administration, RS cells obtained from tumor masses after in vivo treatment, were ex vivo reexposed to drugs and apoptotic responses evaluated. Specifically, we tested RS cells purified from regrown tumor masses of Duv- (supplemental Figure 3A), Ven- (supplemental Figure 3B), or Duv/Ven-treated mice (supplemental Figure 3C) of the RS1316 and RS9737 models. Results indicated that RS cells were still sensitive to the drugs as shown by a marked decrease in cell viability after 24 hours treatment compared with vehicle-treated cells. Both single agents and their combination were capable of inducing significant apoptosis, with the combination showing the best results in both models.
It is noteworthy that these results were similar to the apoptotic rate obtained in RS cells purified from untreated mice and ex vivo exposed to Duv, Ven, or their combination (Figure 2A), suggesting that tumor regrowth is mainly due to treatment discontinuation rather than acquisition of resistance by RS cells.
In vivo combination of Duv and Ven prolongs survival in RS-PDX systemic models
Finally, we moved to a more physiological RS model in which neoplastic cells were IV injected in NSG mice and left to engraft for 10 to 14 days before randomization in 4 groups. Treatment was administered daily for 10 consecutive days, at the doses described previously, and then the mice were monitored for survival and euthanized when reaching clear signs of disease, including cachexia and a weight loss ∼20% of the initial body weight. For this set of experiments, we used the 2 responder PDX models expressing PI3K and Bcl-2 and already adapted to engraft and growth in a systemic way. In line with previous data, RS1316 vehicle-treated mice showed a median survival of 53 days after cell injection.19 Consistent with ex vivo and in vivo results, Kaplan-Meier curves showed prolonged survival in treated mice. Duv or Ven treatment resulted in a 6 days delay compared with vehicle-treated mice (P = .0623 and P = .0101, respectively), whereas their combination further increased the median survival with a total delay compared with vehicle-treated mice of 13 days (P = .0101; Figure 5A). Similar results were obtained with the RS9737 model, with a median survival of vehicle-treated mice of 26 days. Duv and Ven alone slightly increased mice survival with a 5- (P = .0177) and 7- (P = .0177) day delay, respectively, with the dual administration of Duv and Ven resulting in a better response with mice euthanized after 36 days after cell injection (P = .0177; Figure 5B).
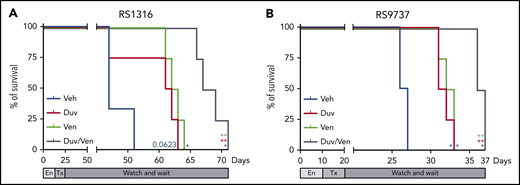
Survival of RS-PDX IV models treated with Duv, Ven, and their combination. Survival curves of (A) RS1316 and (B) RS9737 systemic RS-PDX models treated with vehicle (Veh; blue), Duv (red), Ven (green), and their combination (Duv/Ven; gray). En, engraftment; Tx, treatment. Statistical analysis was performed using Mantel-Cox test; *P < .05, **P < .01, ***P < .001, ****P < .0001.
Survival of RS-PDX IV models treated with Duv, Ven, and their combination. Survival curves of (A) RS1316 and (B) RS9737 systemic RS-PDX models treated with vehicle (Veh; blue), Duv (red), Ven (green), and their combination (Duv/Ven; gray). En, engraftment; Tx, treatment. Statistical analysis was performed using Mantel-Cox test; *P < .05, **P < .01, ***P < .001, ****P < .0001.
GSK3β is at the crossroad between PI3K and Bcl-2 pathways
Last, we focused on the molecular mechanisms at the basis of the synergism between Duv and Ven in RS cells. Several evidences in literature, using different cell types and disease models, suggest a crosstalk between PI3K signaling pathway and the expression of proliferative molecules, such as c-Myc, and antiapoptotic proteins, including Mcl-1, via a concomitant activation of Akt and inactivation of GSK3β, obtained, for both molecules, through their phosphorylation.23-28 The involved mechanism relies on protein ubiquitination and proteasome degradation.
To address this point, we treated RS cells from responder models with Duv, Ven, and their combination and checked for Akt and GSK3β phosphorylation and for Mcl-1 and c-Myc expression. Data indicated that 6 hours’ treatment with Duv resulted in decreased activation of the PI3K signaling pathway, as indicated by lower levels of p-Akt and p-GSK3β, and a diminished expression of Mcl-1 and c-Myc. These effects were further enhanced by the simultaneous administration of Duv and Ven (Figure 6A; supplemental Figure 4A-B). The γ- or δ-selective PI3K inhibitors yielded comparable levels of pathway inhibition and Mcl-1 degradation, underlining that both isoforms are necessary (supplemental Figure 4C).
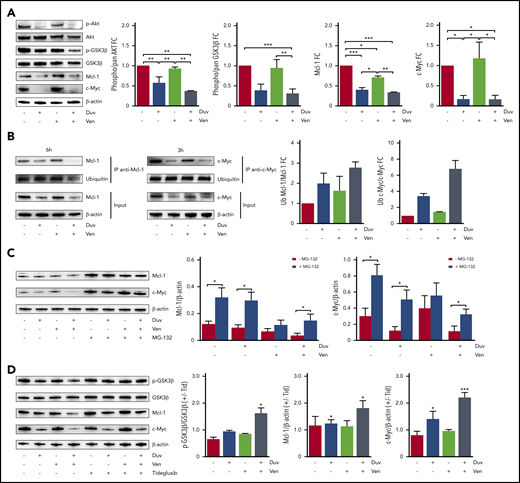
Molecular mechanism underlying the synergism between Duv and Ven in RS cells. (A) Expression levels and activation status of a panel of proteins involved in survival signaling (Akt, GSK3β, Mcl-1, c-Myc) in RS1316 cells after ex vivo treatments (6 hours) with Duv, Ven, their combination, or dimethyl sulfoxide (DMSO). Bar plots represent protein bands intensities (5 independent experiments), measured using Image Lab, normalized on β-actin, and plotted as fold change (FC) over the band of untreated cells. Data are reported as mean ± SEM. (B) Anti-Mcl-1 and anti-c-Myc immunoprecipitation followed by immunoblotting for ubiquitin in RS1316 cells after ex vivo treatments (3 and 6 hours) with Duv, Ven, their combination, or DMSO. Bar plots represent the ratio of ubiquitinated over total protein. (C) Expression levels of Mcl-1 and c-Myc in RS1316 cells after ex vivo treatments (6 and 3 hours, respectively) with Duv, Ven, or their combination in the absence or presence of the proteasome inhibitor MG-132. Band intensities were normalized on β-actin. (D) Expression levels of p-GSK3β, GSK3β, Mcl-1, and c-Myc in RS1316 cells after ex vivo treatments with Duv, Ven, or Duv/Ven in the absence or presence of the GSK3β inhibitor tideglusib. Bar plots represent protein bands intensities (3 independent experiments), measured using Image Lab, normalized on β-actin, and plotted as ratio between tideglusib-treated and untreated samples. Data are reported as mean ± SEM. Statistical analysis was performed using 1-way ANOVA; *P < .05, **P < .01, ***P < .001, ****P < .0001.
Molecular mechanism underlying the synergism between Duv and Ven in RS cells. (A) Expression levels and activation status of a panel of proteins involved in survival signaling (Akt, GSK3β, Mcl-1, c-Myc) in RS1316 cells after ex vivo treatments (6 hours) with Duv, Ven, their combination, or dimethyl sulfoxide (DMSO). Bar plots represent protein bands intensities (5 independent experiments), measured using Image Lab, normalized on β-actin, and plotted as fold change (FC) over the band of untreated cells. Data are reported as mean ± SEM. (B) Anti-Mcl-1 and anti-c-Myc immunoprecipitation followed by immunoblotting for ubiquitin in RS1316 cells after ex vivo treatments (3 and 6 hours) with Duv, Ven, their combination, or DMSO. Bar plots represent the ratio of ubiquitinated over total protein. (C) Expression levels of Mcl-1 and c-Myc in RS1316 cells after ex vivo treatments (6 and 3 hours, respectively) with Duv, Ven, or their combination in the absence or presence of the proteasome inhibitor MG-132. Band intensities were normalized on β-actin. (D) Expression levels of p-GSK3β, GSK3β, Mcl-1, and c-Myc in RS1316 cells after ex vivo treatments with Duv, Ven, or Duv/Ven in the absence or presence of the GSK3β inhibitor tideglusib. Bar plots represent protein bands intensities (3 independent experiments), measured using Image Lab, normalized on β-actin, and plotted as ratio between tideglusib-treated and untreated samples. Data are reported as mean ± SEM. Statistical analysis was performed using 1-way ANOVA; *P < .05, **P < .01, ***P < .001, ****P < .0001.
The reduced expression of Mcl-1 and c-Myc following Duv exposure was dependent on their ubiquitination. Indeed, immunoprecipitation of these targets after Duv treatment, for 3 and 6 hours, in the RS1316 model used because it is the best responder, confirmed their reduced expression and clearly showed that they were heavily ubiquitinated, with the higher ratio between the modified form over the total protein measured in the drugs combination (c-Myc: 6.81 ± 1.446; Mcl-1: 2.774 ± 0.395; Figure 6B). The choice of treating RS cells for different time points before Mcl-1 and c-Myc immunoprecipitation was dictated by the discrepancy in the degradation kinetics of these molecules following drug exposure. Indeed, c-Myc expression was already reduced after 3 hours exposure to Duv, whereas Mcl-1 was degraded starting from 6 hours of treatment (supplemental Figure 5A). In contrast, Ven exposure did not influence Mcl-1, c-Myc, and Bcl-2 expression (supplemental Figure 5A-B). Interestingly, Duv/Ven combination induced a strong downregulation of Bcl-2 expression with a longer kinetics (24 and 48 hours; supplemental Figure 5B). These effects were not due to RS1316 cell death. Indeed, based on live apoptosis monitoring experiments at 3 and 6 hours, drug-treated RS cell viability was similar to vehicle-treated cells, as confirmed by flow cytometry staining (supplemental Figure 5C-D).
As a further demonstration of this molecular mechanism, RS cells were treated with Duv, Ven, and Duv/Ven in the absence or presence of MG-132, a selective proteasome inhibitor. As expected, exposure to this compound resulted in a global accumulation of ubiquitinated proteins compared with the untreated condition (supplemental Figure 5E) with no toxic effect on cells (supplemental Figure 5F). When looking at Mcl-1 and c-Myc expression, MG-132 treatment completely reversed the downregulation induced by Duv and Duv/Ven administration, confirming that proteasome degradation was the underlying pathway (Figure 6C).
Finally, using tideglusib, a non-ATP competitive inhibitor that maintains GSK3β in a phosphorylated and thus inactive form, we showed that Duv/Ven synergism in RS cells was mediated by GSK3β. Indeed, in this condition, both Mcl-1 and c-Myc expression levels were partially restored compared with untreated cells (Figure 6D).
Overall, these data indicate that inhibition of PI3K signaling pathway via Duv results in GSK3β activation, leading to the ubiquitination and subsequent degradation via proteasome of Mcl-1 and c-Myc. The simultaneous targeting of Bcl-2 by Ven further contributes to the induction of apoptosis in RS cells through a coordinated inhibition of antiapoptotic proteins and shut down of proliferative signals (Figure 7).
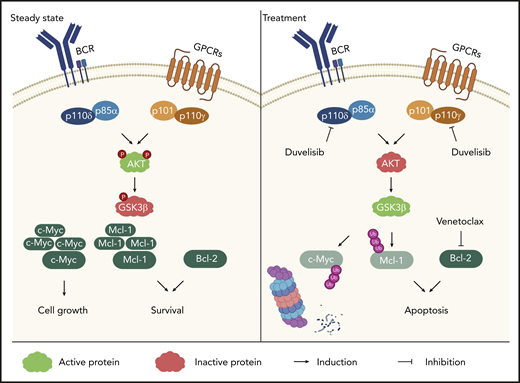
Sketch representing the molecular mechanism beneath Duv and Ven synergism. Molecular pathways activating PI3K-δ/γ signaling (Akt and GSK3β phosphorylation). Following duvelisib treatment, PI3K signaling is inhibited resulting in Mcl-1 and c-Myc ubiquitination and degradation. The simultaneous inhibition of Bcl-2 further sustains RS cell apoptosis.
Sketch representing the molecular mechanism beneath Duv and Ven synergism. Molecular pathways activating PI3K-δ/γ signaling (Akt and GSK3β phosphorylation). Following duvelisib treatment, PI3K signaling is inhibited resulting in Mcl-1 and c-Myc ubiquitination and degradation. The simultaneous inhibition of Bcl-2 further sustains RS cell apoptosis.
Discussion
RS is defined as the development of an aggressive lymphoma from a preexisting CLL. From the clinical perspective, an open point is whether novel targeted therapies, widely used to treat patients with CLL, may affect disease transformation in terms of incidence and selection of more aggressive clones.3 Therapeutic management of patients with RS is still largely unsuccessful. Several clinical trials exploring targeted therapies combined with chemo-immunotherapies are ongoing, and some of them are reporting encouraging results.29,30 Immune therapy based on CD19-directed chimeric antigen receptor-modified T cells has been evaluated as an alternative approach for patients with RS.31 Nevertheless, clinical responses are still partial and limited, with no therapies approved, leaving the disease in an area of high unmet clinical need. Rarity of primary samples and absence of cell lines have so far hampered the ability to study RS biology and to investigate novel therapeutic strategies.
RS-PDX models may represent unique tools to uncover genetic and molecular drivers of the disease and to explore the efficacy of novel therapies.19,20,32 By exploiting these models, we characterized PI3K-γ/δ and Bcl-2 expression and evaluated their therapeutic potential by using targeted drugs. We first examined a cohort of 24 primary samples, obtained from lymph node or bone marrow biopsies of patients with RS. Results obtained by immunohistochemistry staining indicate that PI3K-δ is expressed by primary RS cells, whereas PI3K-γ and Bcl-2 are not homogenous among the cohort. This molecular pattern of expression was confirmed in our RS-PDX models. Although PI3K-δ was invariably expressed by all models at high levels, PI3K-γ was still present but at heterogeneous levels among the models, with RS9737 showing the highest levels. PI3K regulatory subunits, p85α and p101, showed the same expression profile of their respective catalytic subunits. On the contrary, Bcl-2 was abundantly expressed in RS1316, IP867/17, and RS9737, whereas it was completely absent in RS1050. At variance with the other models, this latter one was characterized by a significant expression of the antiapoptotic molecule Mcl-1.
We then analyzed the functional responses of RS cells to PI3K and Bcl-2 targeting by Duv and Ven, 2 drugs currently on the market to treat different leukemias and lymphomas, including CLL. Simultaneous inhibition of PI3K and Bcl-2, using either selective inhibitors or Duv, has shown to be effective in acute myeloid leukemia,33 CLL,34 and DLBCL.27,28,35 However, none of the available studies examined effects in RS patients/models. In addition, at the time this work was started, Duv and Ven combination was under investigation for treatment of relapsed and refractory CLL and small lymphocytic leukemia in a phase 1 clinical trial (NCT03534323), prompting us to evaluate the efficacy of these drugs for RS treatment. It is also noteworthy that this is the only ongoing clinical trial, at least to our knowledge, analyzing the therapeutic response of a PI3K inhibitor combined with Ven.
Ex vivo data indicated that either drug as a single agent presented only limited responses, whereas Duv/Ven had a synergistic effect in inducing apoptosis in 3 of 4 RS-PDX models. In line with the molecular profile, RS1050, characterized by the absence of Bcl-2 and lower levels of PI3K-γ subunit, showed complete resistance to these inhibitors. These data suggest that RS, at variance with DLBCL, the expression of drug molecular targets, may be sufficient to obtain a pharmacological response when cells are exposed to Duv/Ven drug combination. Indeed, in DLBCL, a positive response was usually correlated to BCR dependency and Bcl-2 genetic dysregulation/overexpression, whereas in RS target expression seems to be the limiting factor to predict responses.27,28
When tested in vivo, in an SC model in which RS cells were injected double-flank in mice, Duv and Ven maintained their efficacy, resulting in a significant tumor growth inhibition and induction of apoptosis in RS1316, IP867/17, and RS9737, the responder RS-PDX models. Consistently, RS1050 was fully resistant, excluding also any potential off-target effects of these 2 drugs. Furthermore, efficacy of Duv/Ven combination seemed to be directly correlated to the expression level of drug targets. These results strongly support the hypothesis that patients with RS can be molecular profiled before treatment decision to predict sensitivity to drugs, moving toward a precision medicine approach. In line with this observation, it has already been shown in multiple myeloma models that expression profile of Bcl-2 can predict response to Ven.36
Even when tested in a more physiological model of RS where tumor cells are IV injected and capable of reaching the main lymphoid organs, including spleen and bone marrow, Duv/Ven resulted in a prolonged survival of mice. In both the SC and IV models, mice presented signs of tumor regrowth after drug discontinuation. It is also notable that RS cells were still sensitive to ex vivo drug exposure, suggesting that tumor regrowth was mainly due to treatment discontinuation rather than acquisition of resistance. These results support for the use of Duv/Ven for RS patients’ treatment, even though a better scheduling of their administration is necessary.
Finally, we pinpointed the molecular mechanism underlying the synergism between Duv and Ven in RS. Several lines of evidence in literature suggest a correlation between PI3K signaling pathway and the expression of proliferative molecules, such as c-Myc, and of antiapoptotic proteins, including Mcl-1, via a concomitant activation of Akt and inactivation of GSK3β.27,28,37 We demonstrated that Duv/Ven efficacy in RS cells relies on the concomitant inactivation of Mcl-1, c-Myc, and Bcl-2. Specifically, Duv inhibits PI3K/Akt activation, leading to GSK3β activation, in turn resulting in Mcl-1/c-Myc ubiquitination and degradation via proteasome. Concomitantly, Ven administration inhibits Bcl-2, preventing any balance of Mcl-1 loss and thus dampening the antiapoptotic pathway.
In summary, this work offers preclinical evidence to further investigate the use of Duv and Ven for treatment of patients with RS, and highlights potential for prior molecular profiling to better stratify patients and identify those who will benefit the most from a specific targeted drug treatment.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
For original data, please contact tiziana.vaisitti@unito.it.
Acknowledgments
This work was supported by a Verastem Oncology research grant (S.D.), by the Italian Association for Cancer Research-AIRC (My First AIRC Grant 23107) (T.V.), Investigator grant IG-23095 (S.D.), by the Italian Ministry of Health (GR-2016-02364298) (T.V.), and by the Ministry of Education, University and Research-MIUR “Progetto Strategico di Eccellenza Dipartimentale” (D15D18000410001) (S.D.; as part of the Department of Medical Sciences, University of Turin).
Authorship
Contribution: A.I., S.C., J.A.P., S.D., and T.V. designed research; A.C., A.D.N., J.N.A., and R.R.F. provided material; A.I., N.V., F.A., A.C., A.D.N., and C.L. performed experiments; A.I., S.C., J.A.P., S.D., and T.V. analyzed results and produced figures; A.I., S.C., J.A.P., S.D., and T.V. wrote the manuscript; and all authors approved the final version of the manuscript.
Conflict-of-interest disclosure: J.A.P. and S.C. are employees and shareholders of Verastem Oncology. S.D. has research grants from Verastem Oncology, Heidelberg Pharma and Sunesis. The remaining authors declare no competing financial interests.
Correspondence: Tiziana Vaisitti, Department of Medical Sciences, University of Turin, Via Nizza 52, 10126 Turin, Italy; e-mail: tiziana.vaisitti@unito.it; and Silvia Deaglio, Department of Medical Sciences, University of Turin, Via Nizza 52, 10126 Turin, Italy; e-mail: silvia.deaglio@unito.it.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal