

Key Points
Bcl-2 family members undergo shifts in expression in response to ibrutinib and venetoclax, which revert in case of ibrutinib relapse.
Bcl-XL is dominant over Mcl-1 in CLL venetoclax resistance when both are present.

Abstract
Chronic lymphocytic leukemia (CLL) cells cycle between lymph node (LN) and peripheral blood (PB) and display major shifts in Bcl-2 family members between those compartments. Specifically, Bcl-XL and Mcl-1, which are not targeted by the Bcl-2 inhibitor venetoclax, are increased in the LN. Because ibrutinib forces CLL cells out of the LN, we hypothesized that ibrutinib may thereby affect expression of Bcl-XL and Mcl-1 and sensitize CLL cells to venetoclax. We investigated expression of Bcl-2 family members in patients under ibrutinib or venetoclax treatment, combined with dissecting functional interactions of Bcl-2 family members, in an in vitro model of venetoclax resistance. In the PB, recent LN emigrants had higher Bcl-XL and Mcl-1 expression than did cells immigrating back to the LN. Under ibrutinib treatment, this distinction collapsed; significantly, the pretreatment profile reappeared in patients who relapsed on ibrutinib. However, in response to venetoclax, Bcl-2 members displayed an early increase, underlining the different modes of action of these 2 drugs. Profiling by BH3 mimetics was performed in CLL cells fully resistant to venetoclax due to CD40-mediated induction of Bcl-XL, Mcl-1, and Bfl-1. Several dual or triple combinations of BH3 mimetics were highly synergistic in restoring killing of CLL cells. Lastly, we demonstrated that proapoptotic Bim interacts with antiapoptotic Bcl-2 members in a sequential manner: Bcl-2 > Bcl-XL > Mcl-1 > Bfl-1. Combined, the data indicate that Bcl-XL is more important in venetoclax resistance than is Mcl-1 and provide biological rationale for potential synergy between ibrutinib and venetoclax.
Introduction
Chronic lymphocytic leukemia (CLL) is a B-cell malignancy with considerable heterogeneity in clinical course and a prime example of a malignancy that is highly dependent on interactions with the microenvironment. Whereas quiescent CLL cells accumulate in the peripheral blood (PB), active CLL cells grow in the lymph node (LN), spleen, and bone marrow. In the lymphoid microenvironment, CLL cells are provided with external signals from surrounding cells, such as CD40L-presenting T helper cells,1 myeloid cells, and stromal cells. The result is an upregulation of antiapoptotic Bcl-2 members Bcl-XL, Mcl-1, and Bfl-1, which renders these CLL cells less sensitive to therapeutic drugs.2-4 Novel therapeutics that target these microenvironmental signals or Bcl-2 members have entered clinical practice.1,5
The B-cell receptor (BCR) signaling pathway has emerged as an important therapeutic target for B-cell malignancies, including CLL. Bruton’s tyrosine kinase (BTK), a critical component of BCR signaling, plays a role in the survival, proliferation, adhesion, and migration of CLL cells.6-9 The BTK inhibitor ibrutinib shows strong clinical activity and allows for treatment without chemotherapy. Ibrutinib is now approved for relapsed/refractory and first-line CLL, irrespective of 17p deletions. Importantly, because ibrutinib inhibits CLL cell adhesion and migration, the interaction of CLL cells with their LN microenvironment is blocked, causing rapid redistribution of CLL cells from the LN into the PB. While kept in the circulation, CLL cells are deprived of prosurvival stimuli and will eventually die.10 However, resistance to ibrutinib may occur in a subset of patients, particularly in heavily pretreated patients and in high-risk CLL.11,12 Resistance to ibrutinib correlates with aggressive disease that is difficult to treat and is often associated with acquisition of mutations in primary target BTK or downstream effectors like phospholipase C-γ-2.
Another successful strategy in CLL is to target the apoptosis machinery directly by so-called “BH3 mimetics.” The Bcl-2–specific inhibitor ABT-199, also known as venetoclax, is highly cytotoxic for CLL cells, achieving a 79% overall response rate in relapsed/refractory CLL.13 Venetoclax provides profound reductions in circulating CLL cells in the majority of patients, but LN responses are less complete.13 Although definitive evidence is lacking that residual LN sites after venetoclax treatment are the direct source of resistant disease, it is reasonable to assume that they contribute to this process. Although resistance to ibrutinib has been strongly associated with specific gene mutations in BTK and PLCG2, genetic causes of treatment resistance toward venetoclax, such as the G101V mutation in BCL2, are found in ∼50% of patients under prolonged venetoclax treatment.14 Additional venetoclax-resistance mechanisms, including Bcl-XL overexpression, were observed, showing that alternative mechanisms play a role in venetoclax resistance. Moreover, Blombery et al recently reported the presence of multiple BCL2 mutations, as well as simultaneous aberrations in Bcl-XL and Mcl-1, which further support a model in which multiple independent molecular mechanisms may underlie clinical relapse on venetoclax.15
We hypothesize that changes in expression and shifts in functional interactions among prosurvival Bcl-2 members and their interactions with proapoptotic Bim are key to clinical response and refractory disease. To investigate this, we developed 3 specific objectives.
First, because expression patterns of the different Bcl-2 members play a crucial role in evasion of cell death, which is a hallmark of resistance, it is important to investigate expression changes in response to treatment. Therefore, by measuring the expression of Bcl-2 members in patient samples before and during ibrutinib or venetoclax treatment, we aim to relate Bcl-2 member expression to clinical response.
Second, as outlined above, prosurvival signals in the LN upregulate other Bcl-2 members, such as Bcl-XL and Mcl-1, that are not targeted by venetoclax. Therefore, we analyzed the contribution and interdependency of distinct Bcl-2 members in resistance to venetoclax in CLL in an in vitro CLL model to examine where resistance could be counteracted using a combination of BH3 mimetics targeting other Bcl-2 members, such as Bcl-XL and Mcl-1.
Finally, we aimed to dissect the hierarchy of Bcl-2 members16 in CLL to understand the mechanisms of venetoclax resistance. Therefore, we performed Bim coimmunoprecipitation experiments to investigate shifts in functional interactions of prosurvival Bcl-2 members with proapoptotic Bim in response to treatment to define a hierarchy of prosurvival Bcl-2 members and their relative importance in venetoclax resistance in CLL.
Materials and methods
Patient material
After informed consent, patient blood was obtained during diagnostic or follow-up procedures at the Amsterdam University Medical Centers, the University of Southampton, the Peter MacCallum Cancer Centre and St. Vincent’s Hospital, and the Dana-Farber Cancer Institute. This study was approved by the Academic Medical Center Ethical Review Board and conducted in agreement with the Declaration of Helsinki. The samples from the University of Southampton were collected as part of the ethically approved United Kingdom real-world observational study “A Longitudinal Observational Study with Phenotypic, Functional and Molecular Characterization of the Tumor Lymphoid Cells in Patients with Mature Lymphoid Malignancies in the United Kingdom” (REC reference 16/SC/0030; IRAS ID 186109). The samples from the Peter MacCallum Cancer Centre and St. Vincent’s Hospital were collected under protocol 13/36 as part of the study “A Prospective Study To Evaluate Tumor Biomarkers and Host Immunity in Patients with CLL/SLL,” which was approved on 27 June 2013. At the Dana-Farber Cancer Institute, all patients had previously consented to a serial tissue banking protocol approved by the Institutional Review Board and providing linked clinical information. Blood mononuclear cells of patients with CLL, obtained after Ficoll (Pharmacia Biotech) density gradient centrifugation, were cryopreserved and stored as previously described. Expression of CD5 and CD19 (BD Biosciences, San Jose, CA) on leukemic cells was assessed by flow cytometry using a BD FACSCanto (BD Biosciences). CLL samples included in this study contained 85% to 99% CD5+/CD19+ cells.
Reagents
ABT-199 was purchased from Active Biochem (Bonn, Germany), A-1331852 was purchased from ChemieTek (Indianapolis, IN), S-63845 was purchased from Chemgood (Glen Allen, VA), and Q-VD-OPh hydrate was purchased from APExBIO (Houston, TX).
Cell culture and detection of apoptosis
Lymphocytes of CLL patients were cocultured with NIH3T3 fibroblasts stably transfected with human CD40L or negative control, as described previously.17,18 Twenty-four hours later, CLL cells were detached and incubated, with or without drugs, for an additional 24 hours. CLL cell viability was measured by flow cytometry using DiOC6 and TO-PRO-3 viability dyes. Specific apoptosis is defined as (% cell death in treated cells) − (% cell death in medium control)/(% viable cells medium control) × 100.
Flow cytometry
Single-cell suspensions were stained with the following antibodies: anti-CD3, anti-CD19 (BD Biosciences), ant-iCD184 (CXCR4), and anti-CD5 (BioLegend). Cells were subsequently permeabilized and stained with the following antibodies: anti–Bcl-2 (BioLegend), anti–Bcl-XL, and anti–Mcl-1 (Cell Signaling Technology). Stained cells were analyzed on a BD FACSCanto II cytometer (BD Biosciences). Gating of LN emigrants vs immigrants was performed on plots of CD19+/CD5dim/high/CXCR4dim/high populations and setting the optimal 10 percentiles corresponding to the 2 populations. The proportions of immigrants and emigrants shifted in some patients during treatment, as has been reported previously,19 but this did not affect interpretation of the gating strategy (supplemental Figure 1, available on the Blood Web site).
Western blot analysis
Western blot analysis was performed using standard techniques. Membranes were probed with the following antibodies: anti–Bcl-XL, anti–Bcl-2 (Cell Signaling), anti–Mcl-1 (Abcam), anti-actin (Santa Cruz Biotechnology), anti-Bim (StressMarq Biosciences), and anti-A1/Bfl-1 (gift from J. Borst, The Netherlands Cancer Institute, Amsterdam, The Netherlands). An Odyssey Imager (Li-Cor Biosciences) was used as a detection method for the signal of the secondary western blot antibodies at the 680 nm and 800 nm wavelengths, according to the manufacturer’s protocol. When PB samples contained <90% CLL cells, they were purified using CD19 beads (Miltenyi Biotec) (data not shown).
Bim coimmunoprecipitation
Lymphocytes of CLL patients were cocultured with NIH3T3 fibroblasts stably transfected with human CD40L or cocultured with negative-control NIH3T3 fibroblasts transfected with empty vector. After 24 hours, CLL cells were detached and incubated, with or without BH3 mimetics, in combination with caspase inhibitor QVD for an additional 24 hours. Treated CLL cells were lysed with NP-40 lysis buffer and subjected to immunoprecipitation using a Dynabeads Protein G Immunoprecipitation Kit (catalog no. 10007D; Thermo Fisher Scientific). Rat anti-Bim (3C5) monoclonal antibodies were coupled to Protein G Dynabeads in a 1:50 dilution for 20 minutes on a rotator at room temperature. NP-40 lysates were incubated with antibody-coated beads for 20 minutes on a rotator at room temperature. All other steps were performed according to the manufacturer’s protocol.
Statistics
A paired-sample Student t test was used to analyze paired observations. One-way analysis of variance was used to analyze differences between groups.
Results
Levels of Bcl-XL and Mcl-1 collapse in LN emigrants upon ibrutinib treatment
CLL cells can be divided into distinct fractions based on the inverse surface expression of CXCR4 and CD5. Separation and intraclonal analyses of these fractions in PB samples suggest a spectrum of CLL cells from recently divided and robust cells that are recent lymphoid tissue emigrants (CXCR4dim/CD5high) to older and less vital cells that need to immigrate back to lymphoid tissue to survive (CXCR4high/CD5dim).20 Because CLL cells upregulate Mcl-1 and Bcl-XL in the LN,3,4 we hypothesized that LN emigrants have higher Mcl-1 and Bcl-XL expression than LN immigrants. To this end, we analyzed PB samples; no other compartments were studied. By gating the outer 10% fractions corresponding to these 2 populations, CLL cells emigrating from the LN indeed showed higher Mcl-1 and Bcl-XL expression than did CLL cells immigrating back to the LN (Figure 1A; supplemental Figure 2). Under ibrutinib treatment, levels of Mcl-1 and Bcl-XL in the LN emigrant population gradually decreased to the levels of the LN immigrant population. In some patients, the proportion of the immigrant vs emigrant population shifted upon ibrutinib treatment over time, as has recently been reported,19 whereas in other patients this appeared to be stable over the period examined. Relative pairwise comparisons were made by setting the expression levels of the LN immigrant population at baseline to 100% (see supplemental Figure 1 for an example). This demonstrated a significant reduction in the LN emigrant population of Mcl-1 and Bcl-XL upon ibrutinib treatment (n = 14). Bcl-2 levels were affected in some patients; however, on average, the changes were not significant (Figure 1B). These findings were not influenced by previous treatment regimens (supplemental Table 1). Although the difference in Bcl-2 family proteins is expected and observed to be the greatest between the LN immigrant and emigrant populations, as gated by flow cytometry, these findings could, in some cases, be confirmed in the total fraction of CLL cells via western blotting of PB samples from patients undergoing ibrutinib treatment. The data obtained by western blot technique were compared with flow cytometric data for the total CLL population obtained after CD40 stimulation in selected cases and proved to correlate well (supplemental Figure 3). However, it should be pointed out the western blot technique, which analyzes the entire CLL population, cannot be expected to show the same clear differences as the specific LN immigrant and emigrant populations shown in the flow cytometric analyses. Reductions in Mcl-1 and Bcl-XL levels were observed in patients upon short-term (Figure 1C) and long-term (Figure 1D) ibrutinib treatment compared with baseline samples (supplemental Figures 4-5). In summary, levels of Bcl-XL and Mcl-1, which are known to be responsive to microenvironmental signals, collapse in response to ibrutinib treatment in PB CLL cells, indicating that the lack of microenvironmental signals extends into that compartment.
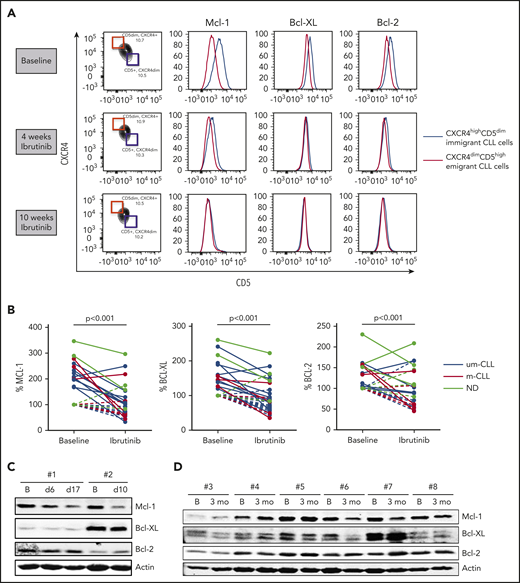
Levels of Bcl-2 members collapse upon ibrutinib treatment. (A) FACS analysis showing overlays of Mcl-1, Bcl-XL, and Bcl-2 expression for LN immigrants and emigrants. Data are from a representative patient at baseline and after 4 and 10 weeks of ibrutinib treatment. (B) Summary of Bcl-2 member expression in LN emigrants before and after ibrutinib treatment (n = 14). Geometric mean fluorescence intensities were normalized by setting the baseline LN immigrant population at 100%. Black lines indicate unmutated IgHV CLL samples (um-CLL), red lines indicate mutated IgHV CLL samples (m-CLL), and gray lines indicate patient samples for whom the mutation status was not determined (ND). A paired-sample Student t test was used for statistical analyses. (C-D) Western blots of peripheral blood collected from 8 patients treated with ibrutinib. Samples were collected at baseline and after 6 to 17 days of ibrutinib treatment (patients #1 and #2) or after 3 months of ibrutinib treatment (patients #3-8). Protein lysates were probed for Mcl-1, Bcl-XL, and Bcl-2; actin was used as loading control. B, baseline; ns, not significant.
Levels of Bcl-2 members collapse upon ibrutinib treatment. (A) FACS analysis showing overlays of Mcl-1, Bcl-XL, and Bcl-2 expression for LN immigrants and emigrants. Data are from a representative patient at baseline and after 4 and 10 weeks of ibrutinib treatment. (B) Summary of Bcl-2 member expression in LN emigrants before and after ibrutinib treatment (n = 14). Geometric mean fluorescence intensities were normalized by setting the baseline LN immigrant population at 100%. Black lines indicate unmutated IgHV CLL samples (um-CLL), red lines indicate mutated IgHV CLL samples (m-CLL), and gray lines indicate patient samples for whom the mutation status was not determined (ND). A paired-sample Student t test was used for statistical analyses. (C-D) Western blots of peripheral blood collected from 8 patients treated with ibrutinib. Samples were collected at baseline and after 6 to 17 days of ibrutinib treatment (patients #1 and #2) or after 3 months of ibrutinib treatment (patients #3-8). Protein lysates were probed for Mcl-1, Bcl-XL, and Bcl-2; actin was used as loading control. B, baseline; ns, not significant.
Reappearance of Bcl-2 members upon ibrutinib relapse
We next investigated whether Bcl-2 member expression would reappear in patients relapsing on ibrutinib therapy. PB samples were collected at multiple time points during ibrutinib treatment where T1 represents an early time point and T2 represents a later time point upon ibrutinib treatment (supplemental Table 1). In this group of patients, significant reductions in all Bcl-2 members were observed in the LN emigrant population upon ibrutinib treatment (Figure 2A) (n = 16). Subsequently, patients were divided into remission (n = 7) and nonresponding/relapsing (n = 9) groups. At T2, Mcl-1 and Bcl-XL expression was significantly different between patients in remission vs nonresponding/relapsing patients. When sufficient material was available, Bcl-2 members were analyzed by western blot (Figure 2B; supplemental Figure 6). Although the increase in Mcl-1 and Bcl-XL expression between T1 and T2 was variable between patients, their expression for each given patient was correlated (supplemental Figure 7). All nonresponding/relapsing patients were sequenced for mutations in BTK and PLCG2; 1 patient contained a BTK mutation. In summary, these observations show that Bcl-XL and Mcl-1 levels increase upon ibrutinib relapse, irrespective of the distinct time points when relapse occurred.
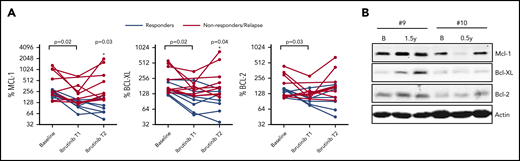
Reappearance of Bcl-2 members upon ibrutinib relapse. (A) Summary of Bcl-2 member expression in LN emigrants before and during ibrutinib treatment (n = 16). Ibrutinib T1 represents an early time point showing clinical response to ibrutinib, and T2 represents a later time point upon ibrutinib treatment. Red lines indicate patients who relapsed on ibrutinib (n = 9), and black lines indicate patients who stayed in remission (n = 7). Geometric mean fluorescence intensities were normalized by setting the baseline LN immigrant population at 100%. The paired-sample Student t test was used for statistical analysis for all patients to compare expression at baseline with expression upon ibrutinib treatment. Subsequently, patients were divided into remission or relapse groups, after which 1-way analysis of variance was used to compare Bcl-2 member expression between groups during follow-up. (B) Western blot of peripheral blood collected from 2 patients who relapsed on ibrutinib. Samples were collected at baseline, after 0.5 to 1.5 years of ibrutinib treatment, and during relapse on ibrutinib after 4 years (patient #9) and 1 year (patient #10) of treatment. Protein lysates were probed for Mcl-1, Bcl-XL, and Bcl-2; actin was used as loading control. B, baseline.
Reappearance of Bcl-2 members upon ibrutinib relapse. (A) Summary of Bcl-2 member expression in LN emigrants before and during ibrutinib treatment (n = 16). Ibrutinib T1 represents an early time point showing clinical response to ibrutinib, and T2 represents a later time point upon ibrutinib treatment. Red lines indicate patients who relapsed on ibrutinib (n = 9), and black lines indicate patients who stayed in remission (n = 7). Geometric mean fluorescence intensities were normalized by setting the baseline LN immigrant population at 100%. The paired-sample Student t test was used for statistical analysis for all patients to compare expression at baseline with expression upon ibrutinib treatment. Subsequently, patients were divided into remission or relapse groups, after which 1-way analysis of variance was used to compare Bcl-2 member expression between groups during follow-up. (B) Western blot of peripheral blood collected from 2 patients who relapsed on ibrutinib. Samples were collected at baseline, after 0.5 to 1.5 years of ibrutinib treatment, and during relapse on ibrutinib after 4 years (patient #9) and 1 year (patient #10) of treatment. Protein lysates were probed for Mcl-1, Bcl-XL, and Bcl-2; actin was used as loading control. B, baseline.
Levels of Bcl-2 members increase upon venetoclax treatment
The same analyses were applied to samples from patients treated with venetoclax. Samples were collected twice during the ramp-up phase when the dosage of venetoclax is gradually increased and there are still detectable CLL cells present in the PB (supplemental Figure 8). Significant increases in all Bcl-2 members could be observed in LN immigrants and emigrants after the first sampling, with further increases after the second sampling (Figure 3A). Samples were also compared by western blot (Figure 3B; enriched for CD19+ cells to achieve >95% purity of CD5+CD19+ CLL cells). Upon 2 weeks of venetoclax ramp-up, an increase in Bcl-2 could be observed in all patients, whereas an increase in Mcl-1 and Bcl-XL could be observed when PB was collected 4 weeks after starting the venetoclax ramp-up (supplemental Figures 9-10). Quantitative polymerase chain reaction analyses to probe for changes in RNA expression of the various Bcl-2 members showed incidental and varying changes but not a consistent pattern (data not shown). To establish whether this venetoclax-associated increase in Bcl-2 member protein levels in vivo was due to short-term effects on Bcl-2 members, CD40-stimulated CLL cells were treated in vitro with venetoclax (ABT199) and other BH3 mimetics targeting Mcl-1 (S-63845) or Bcl-XL (A-1331852) (Figure 3C). Upon CD40-mediated activation of CLL cells using a coculture of CLL cells with CD40L-expressing 3T3 fibroblasts (3T40L),21,22 upregulation of Mcl-1 and Bcl-XL could be observed, similar to the situation in the LN.4 Only for Mcl-1 could the in vivo effect be reproduced after in vitro incubation with venetoclax or S-63845. Mcl-1 is unstable and known to be stabilized at the protein level by BH3 protein binding,23-25 suggesting that the observed increase in Mcl-1 after venetoclax treatment may reflect its binding to Bim in vivo. Altogether, these results show that levels of Mcl-1, Bcl-XL, and Bcl-2 increase upon venetoclax treatment.
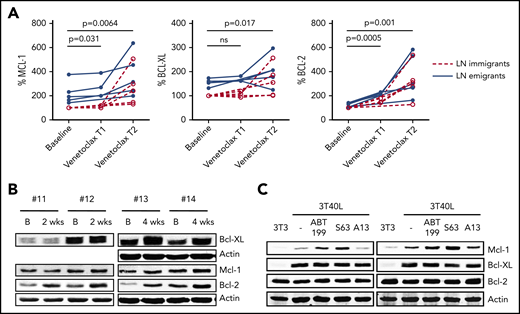
Levels of Bcl-2 members increase upon venetoclax treatment. (A) Summary of Bcl-2 member expression in LN emigrants and immigrants before and after venetoclax treatment (n = 5). Venetoclax T1 and T2 represent time points 1 and 2 (ie, the first and second sampling of PB during venetoclax ramp-up; median time of 3 weeks in between time points). A paired-sample Student t test was used for statistical analyses between baseline and Venetoclax T1 or T2. (B) Western blot of PB collected from 4 patients treated with venetoclax. Samples were collected at baseline and upon 2 to 4 weeks of venetoclax treatment. Protein lysates were probed for Mcl-1, Bcl-XL, and Bcl-2; actin was used as loading control. (C) CLL cells of 2 patients were cultured on 3T3 or 3T40L for 24 hours. After detachment, cells were treated with the BH3 mimetics venetoclax (ABT199), S-63845 (S63), or A-1331852 (A13) for an additional 24 hours. Protein lysates were probed for Mcl-1, Bcl-XL, and Bcl-2; actin was used as loading control. B, baseline; ns, not significant.
Levels of Bcl-2 members increase upon venetoclax treatment. (A) Summary of Bcl-2 member expression in LN emigrants and immigrants before and after venetoclax treatment (n = 5). Venetoclax T1 and T2 represent time points 1 and 2 (ie, the first and second sampling of PB during venetoclax ramp-up; median time of 3 weeks in between time points). A paired-sample Student t test was used for statistical analyses between baseline and Venetoclax T1 or T2. (B) Western blot of PB collected from 4 patients treated with venetoclax. Samples were collected at baseline and upon 2 to 4 weeks of venetoclax treatment. Protein lysates were probed for Mcl-1, Bcl-XL, and Bcl-2; actin was used as loading control. (C) CLL cells of 2 patients were cultured on 3T3 or 3T40L for 24 hours. After detachment, cells were treated with the BH3 mimetics venetoclax (ABT199), S-63845 (S63), or A-1331852 (A13) for an additional 24 hours. Protein lysates were probed for Mcl-1, Bcl-XL, and Bcl-2; actin was used as loading control. B, baseline; ns, not significant.
Functional profiling with BH3 mimetics to dissect venetoclax resistance
Upon starting venetoclax treatment, CLL cells rapidly disappear from the PB.13 Because CLL cells upregulate Bcl-XL, Mcl-1, and Bfl-1 in the lymphoid microenvironment,3,4 these stimulated CLL cells are theoretically less sensitive to single Bcl-2 inhibition by venetoclax treatment. Because this population could potentially survive under prolonged therapy, this may result in outgrowth of venetoclax-resistant subclones. This premise fits with the observation that patients with remaining enlarged LNs under venetoclax treatment have a shorter progression-free survival,13 even when combined with rituximab.26 To dissect the functional contribution of other prosurvival Bcl-2 members to venetoclax resistance, we applied functional profiling using combinations of BH3 mimetics27 targeting Bcl-2 (ABT199), Bcl-XL (A-1331852), and/or Mcl-1 (S-63845) in an in vitro model in which CLL cells are stimulated via coculture with CD40L-expressing fibroblasts (as shown in Figure 3C) and become fully resistant to single-agent venetoclax.27,28 Combined inhibition of Bcl-2 and Bcl-XL (Figure 4A) or Mcl-1 (Figure 4B) diminished resistance. Targeting Bcl-XL seemed slightly more effective than targeting Mcl-1, resulting in a fourfold difference in the 50% inhibitory concentration between the 2 combinations (see supplemental Table 2). Interestingly, combined Mcl-1/Bcl-XL inhibition in the absence of venetoclax was also able to restore killing (Figure 4C). Triple combination of BH3 mimetics in 3T40-stimulated CLL almost completely restored killing comparable to unstimulated PB CLL cells (Figure 4D). However, a further reduction in the dose of the triple combination showed that there was still a difference in sensitivity between CD40-activated CLL and control cells (Figure 4E). Because Bcl-2, Bcl-XL, and Mcl-1 are inhibited in this situation, a role for Bfl-1, which is also induced after CD40 stimulation,2 is implied. Results were similar between mutated and unmutated CLL samples, and 50% inhibitory concentration values were calculated using a nonlinear regression model. The combination index of the different BH3 mimetic combination treatments was calculated (supplemental Figure 11) and showed clear synergy between the dual and, especially, triple combinations (Figure 4F). Altogether, these data demonstrate that full resistance against single-BH3 mimetic drugs after CD40 stimulation can be significantly reversed by dual-BH3 mimetic combinations and that targeting Bcl-XL contributes strongly to this reversion.

BH3 mimetic profiling to dissect venetoclax resistance. CLL cells were cultured on 3T3 or 3T40L for 24 hours. After detachment, cells were treated with the indicated BH3 mimetics for an additional 24 hours. The respective drug concentrations were determined in preliminary experiments. Viability was measured by flow cytometry using DiOC6/TO-PRO-3 staining. Summary of viability data for CD40-stimulated CLL cells treated with BH3 mimetics (n = 16) (A-D) or with low-dose BH3 mimetics (n = 5) (E). (F) Table showing synergy indexes of BH3 mimetics in combination treatments (n = 16).
BH3 mimetic profiling to dissect venetoclax resistance. CLL cells were cultured on 3T3 or 3T40L for 24 hours. After detachment, cells were treated with the indicated BH3 mimetics for an additional 24 hours. The respective drug concentrations were determined in preliminary experiments. Viability was measured by flow cytometry using DiOC6/TO-PRO-3 staining. Summary of viability data for CD40-stimulated CLL cells treated with BH3 mimetics (n = 16) (A-D) or with low-dose BH3 mimetics (n = 5) (E). (F) Table showing synergy indexes of BH3 mimetics in combination treatments (n = 16).
Functional hierarchy among Bcl-2 members in determining resistance to venetoclax
Of the proapoptotic BH3-only proteins expressed in CLL, Bim is expected to play a major part in killing cells upon venetoclax treatment.28 Therefore, Bim was immunoprecipitated from primary CLL cells treated with BH3 mimetics to observe shifts in its interactions with antiapoptotic Bcl-2 proteins (Figure 5A). These experiments were performed in the presence of the pan-caspase inhibitor QVD to prevent cell death. In resting CLL cells, in which Bcl-2 is the most prominent prosurvival Bcl-2 member, Bim only interacted with Bcl-2 (Figure 5B). Upon CD40-mediated activation and subsequent upregulation of Bcl-XL and Mcl-1, Bim was also bound to Bcl-XL. When cells were then treated with venetoclax, Bim shifted further to Mcl-1, confirming that it has a preference for Bcl-XL over Mcl-1 when both are present. Based on data from Figure 4E, it was predicted that Bfl-1 could also play a role; this was confirmed using the triple combination of BH3 mimetics (Figure 5C). Under these conditions, Bim abrogated binding to Bcl-XL and Mcl-1 and switched back to Bcl-2 and now also engaged Bfl-1 (Figure 5D). Although venetoclax was present in this condition, Bcl-2 was still capable of binding Bim when Bcl-XL and Mcl-1 were blocked by other BH3 mimetics. Although Bcl-2 family member expression differed from patient to patient, a similar pattern was found in multiple patient samples (supplemental Figures 12-13). In samples with high Bcl-XL expression, interactions with Bim could already be observed without 3T40 activation. In samples with high Mcl-1 expression, interactions with Bim could still be observed, even after using the triple combination of BH3 mimetics. Finally, Bfl-1 could not always be observed in the coimmunoprecipitation. Overall, these data show that Bim interacts with antiapoptotic Bcl-2 members in a sequential manner in the order of Bcl-2, Bcl-XL, Mcl-1, and, lastly, Bfl-1, revealing a functional hierarchy of antiapoptotic Bcl-2 members in determining resistance to venetoclax.
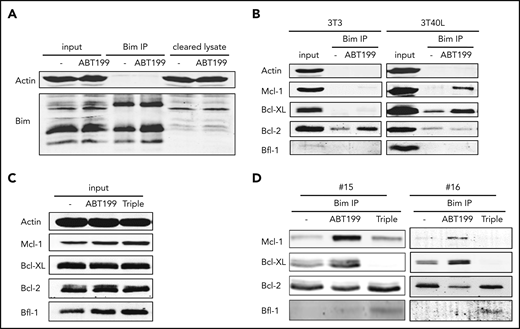
Functional hierarchy of Bcl-2 members in determining resistance to venetoclax. (A-B) CLL cells were cultured on 3T3 or 3T40L for 24 hours. After detachment, cells were treated with 5 µM QVD and 100 nM venetoclax for an additional 24 hours and then lysed with NP-40 lysis buffer. Shown are the protein lysates (input), the proteins in the lysates that were pulled down (Bim IP), and the proteins still present in the lysates after the IP (cleared lysate) (A), as well as the coimmunoprecipitated proteins of the Bim IP (B). (C-D) CLL cells were cultured on 3T40L for 24 hours. After detachment, cells were left untreated (-) or treated with 5 µM QVD and 100 nM venetoclax (ABT199) supplemented or not with 100 nM S-63845 and 300 nM A-1331852 (Triple) for 24 hours before lysis. Shown are the protein lysates (input) (C) and the coimmunoprecipitated proteins of the Bim IP from patients #15 and #16 (D). IP, immunoprecipitation.
Functional hierarchy of Bcl-2 members in determining resistance to venetoclax. (A-B) CLL cells were cultured on 3T3 or 3T40L for 24 hours. After detachment, cells were treated with 5 µM QVD and 100 nM venetoclax for an additional 24 hours and then lysed with NP-40 lysis buffer. Shown are the protein lysates (input), the proteins in the lysates that were pulled down (Bim IP), and the proteins still present in the lysates after the IP (cleared lysate) (A), as well as the coimmunoprecipitated proteins of the Bim IP (B). (C-D) CLL cells were cultured on 3T40L for 24 hours. After detachment, cells were left untreated (-) or treated with 5 µM QVD and 100 nM venetoclax (ABT199) supplemented or not with 100 nM S-63845 and 300 nM A-1331852 (Triple) for 24 hours before lysis. Shown are the protein lysates (input) (C) and the coimmunoprecipitated proteins of the Bim IP from patients #15 and #16 (D). IP, immunoprecipitation.
Discussion
We report the first analyses, in CLL, of the effects of ibrutinib and venetoclax ex vivo and in vitro on prosurvival Bcl-2 members. Significant and opposite effects were found for Bcl-2 members in response to ibrutinib compared with venetoclax. Furthermore, we dissected the contribution of the Bcl-2 members in venetoclax resistance and found a dominant role for Bcl-XL and a functional hierarchy in binding Bim.
As a result of the microenvironment-driven upregulation of Mcl-1 and Bcl-XL in the LN2-4 and the absence of such signals in the PB,20 LN emigrants and immigrants differ in their expression of these Bcl-2 members. Because ibrutinib forces CLL cells out of the LN into the PB and prevents their return, we hypothesized that the difference in Bcl-2 expression levels between LN emigrants and immigrants would collapse upon treatment. Gating specifically for LN immigrants and emigrants by flow cytometry showed a significant decrease of Bcl-XL and Mcl-1 expression upon ibrutinib treatment, whereas these effects could not always be reproduced by blotting total PB. Even though these 2 separate techniques show slightly different aspects, the available data showed that the decreased expression mediated by ibrutinib was true for Bcl-XL and Mcl-1.
Although previous studies did not find consistent differences in the expression of Bcl-XL and Mcl-1 upon ibrutinib therapy,29 this may be explained by the different techniques used, as well as the duration of ibrutinib treatment. No significant differences were observed for Bcl-2, which fits with the notion that Bcl-2 is overexpressed independently of the microenvironment in CLL. Yet, some patients studied here displayed a decrease in Bcl-2 upon ibrutinib treatment, although this did not correspond to IgHV mutation status, as suggested in a preliminary report.30 It is hoped that further evaluation in larger patient cohorts will clarify which environmental or intrinsic signals may affect Bcl-2 upon ibrutinib treatment. Significantly, the Bcl-2 member pretreatment profile reappeared in patients who relapsed on ibrutinib. Mcl-1 and Bcl-XL increased, most likely as the result of CLL cells being able to recharge again in the LN, whereas the increase in Bcl-2 may be due to BTK inhibition initially selecting clones expressing higher levels of Bcl-2.30 Notably, of the 9 relapsing patients who were included, only 1 patient had a BTK mutation, indicating that mechanisms other than BTK or PLCG mutations were involved with the reappearance of Bcl-2 member expression during relapse in this group of patients.
In contrast to ibrutinib, venetoclax induced an increase in the Bcl-2 members Mcl-1, Bcl-XL, and Bcl-2. Mcl-1 also increased upon venetoclax or S-63845 treatment in vitro (Figure 3C), and we propose that Mcl-1 is not upregulated, but rather is stabilized, upon interaction with Bim or S-63845, which are previously reported effects.23-25,31 Increases in Bcl-2 or Bcl-XL are most likely not due to stabilization, because they have a much longer half-life than Mcl-1 and Bfl-1.32 Notably, an increase in Bcl-2 or Bcl-XL was not observed after short-term treatment in vitro. The apparent increase in Bcl-2 may be a result of preferential elimination of cells with low Bcl-2 protein during venetoclax ramp-up. Apart from a theoretical possibility of longer half-life upon binding Bim, the increase in Bcl-XL in vivo remains unexplained, but it may be a potential resistance mechanism. Indeed, increased Bcl-XL expression has been described as a mechanism of resistance to venetoclax in CLL and mantle cell lymphoma.33,34 In addition, Blombery et al identified the G101V Bcl-2 mutation in a cohort of CLL patients treated with venetoclax and showed that additional disease-resistance mechanisms, such as high Bcl-XL expression, can coexist with the G101V Bcl-2 mutation.14 To establish the potential of Bcl-2 member expression as a biomarker for (non)response in the treatment of ibrutinib and/or venetoclax, patients should be tested in a prospective study in correlation with response parameters, such as MRD status, in trials involving venetoclax.35
Using an in vitro CLL model of CD40 stimulation, it was shown that CLL cells with upregulation of Bcl-XL, Mcl-1, and Bfl-1 are fully resistant to single-agent venetoclax.22 We demonstrate that dual or triple targeting with BH3 mimetics restored efficient killing of 3T40-activated CLL cells. Triple combination of BH3 mimetics restored killing of 3T40-activated CLL cells to almost the same level of PB CLL cells. Although such combinations may not be directly translatable to clinical applications, they allow assessment of the contributions of the various Bcl-2 members in CLL cells to venetoclax resistance. In a recent study with primary AML cells, synergy between Bcl-2 and Mcl-1 was observed.36 Previous research, predominantly performed with cell lines, has also suggested that Mcl-1 can play a prominent role in determining resistance to venetoclax or navitoclax (ABT-737).37-41 A recent study using loss- and gain-of-function screens in cell lines also confirmed a prominent contribution of Mcl-1 in venetoclax resistance.42 Previous data using BCR-stimulated CLL cells also indicated a role for Mcl-1 in lowering venetoclax sensitivity.43 Our data reported here, using BH3 mimetics and Bim interaction studies, as well as previous small interfering RNA approaches,22 indicate that, in primary CLL cells, Bcl-XL plays a more prominent role in venetoclax resistance than does Mcl-1 when both are upregulated following CD40 stimulation. Combined, the available data establish that Bcl-XL and Mcl-1 can confer resistance to venetoclax and can be expected to be selected for overexpression during prolonged venetoclax treatment. Because direct targeting of multiple prosurvival Bcl-2 members by combinations of BH3 mimetics in patients may result in toxicity, future studies should be aimed at dissecting the signaling pathways that contribute to venetoclax resistance in relapsing patients. Combined, the BH3 mimetic and Bim interaction studies allow us to establish a hierarchy of Bcl-2 members in binding to Bim: Bcl-2 > Bcl-XL > Mcl-1 > Bfl-1, when all 4 are present. When all available BH3 mimetics were applied, Bim interacted with Bfl-1, confirming that Bfl-1 can also bind Bim in primary leukemic cells and, thus, might play a role in venetoclax resistance in CLL.2,44
Our data are compatible with combination treatment with ibrutinib and venetoclax, which is currently being tested in trials, because ibrutinib forces CLL cells out of the LN, which sensitizes these cells to venetoclax as levels of Mcl-1 and Bcl-XL collapse.
Data sharing requests should be sent to Eric Eldering (e.eldering@amsterdamumc.nl).
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Fabio Brocco for assistance with the experiments shown in Figure 4.
This work was supported by a grant from the European Research Area Network (ERA-NET) on Translational Cancer Research (TRANSCAN 2) in support of the consortium Fighting Resistance in CLL. M.H. was funded by Blaauboer Fund via the AMC Foundation. K.K. was financed by the PPP Allowance made available by Top Sector Life Sciences & Health under project number LSHM18055-SGF. For more information: www.target-to-b.nl.
Authorship
Contribution: M.V.H. performed research, analyzed data, and wrote the manuscript; K.K., J.t.B., and D.J.C.B. performed research and analyzed data; S.M.F. provided patient samples; J.B. provided vital reagents and read and edited the manuscript; C.T., F.F., and J.R.B. provided patient samples and read and edited the manuscript; G.C. and J.D. analyzed patient clinical characteristics; A.P.K. provided patient samples and wrote the manuscript; and E.E. designed the research and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Eric Eldering, Department of Experimental Immunology, Amsterdam University Medical Centers, Location AMC, Meibergdreef 9, 1105AZ Amsterdam, The Netherlands, e-mail: e.eldering@amsterdamumc.nl.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal