

Key Points
PF4 improves outcome in sepsis by stabilizing NETs and enhancing NET-mediated microbial entrapment.
An Fc-modified HIT-like antibody further enhances the beneficial effects of PF4 in sepsis.

Abstract
Sepsis is characterized by multiorgan system dysfunction that occurs because of infection. It is associated with high morbidity and mortality and is in need of improved therapeutic interventions. Neutrophils play a crucial role in sepsis, releasing neutrophil extracellular traps (NETs) composed of DNA complexed with histones and toxic antimicrobial proteins that ensnare pathogens, but also damage host tissues. At presentation, patients often have a significant NET burden contributing to the multiorgan damage. Therefore, interventions that inhibit NET release would likely be ineffective at preventing NET-based injury. Treatments that enhance NET degradation may liberate captured bacteria and toxic NET degradation products (NDPs) and likely be of limited therapeutic benefit as well. We propose that interventions that stabilize NETs and sequester NDPs may be protective in sepsis. We showed that platelet factor 4 (PF4), a platelet-associated chemokine, binds and compacts NETs, increasing their resistance to DNase I. We now show that PF4 increases NET-mediated bacterial capture, reduces the release of NDPs, and improves outcome in murine models of sepsis. A monoclonal antibody KKO which binds to PF4-NET complexes, further enhances DNase resistance. However, the Fc portion of this antibody activates the immune response and increases thrombotic risk, negating any protective effects in sepsis. Therefore, we developed an Fc-modified KKO that does not induce these negative outcomes. Treatment with this antibody augmented the effects of PF4, decreasing NDP release and bacterial dissemination and increasing survival in murine sepsis models, supporting a novel NET-targeting approach to improve outcomes in sepsis.
Introduction
Sepsis is a dysregulated response to infection that leads to life-threatening organ damage. It affects millions of people each year and remains one of the most common causes of mortality worldwide.1,2 Care consists of antibiotics and supportive measures,3 but these interventions do not target the host response that causes much of the morbidity in sepsis,4 and the poor survival rate has not changed significantly for several decades.5 New emphasis is being placed on the identification of novel targeted interventions,6 and the innate immune response has become an area of interest.7 Neutrophils, the most abundant circulating white blood cell,8 play a crucial role in sepsis. They are recruited to inflamed vessels, where they release neutrophil extracellular traps (NETs), webs of negatively charged cell-free DNA (cfDNA) complexed with positively charged histones and antimicrobial proteins, such as myeloperoxidase (MPO) and neutrophil elastase, which ensnare pathogens, limit bacterial spread,9 and degrade inflammatory cytokines.10,11 However, these positive effects occur at the expense of collateral tissue damage. NETs are lysed by circulating DNases,12 releasing NET degradation products (NDPs; eg, cfDNA, MPO-cfDNA complexes, histones) that exert harmful effects. For example, cfDNA triggers the contact pathway13 and fixes complement.14 MPO induces oxidative tissue damage.15 neutrophil elastase cleaves tissue factor pathway inhibitor promoting thrombosis,16 whereas histones cause platelet activation17 and are toxic to endothelial cells.18 In septic patients, plasma levels of NDPs correlate with end-organ damage and mortality.19,20 It has been suggested that preventing NET release (NETosis) might be beneficial,21 but we believe that this strategy may be ineffective because many septic patients have a large amount of NETs formed at the time they present. The use of DNase I to accelerate NET degradation has been proposed as a therapeutic intervention.22 However, results of prior studies with this intervention have been mixed,23 raising the concern that DNase I treatment may induce harm through the release of entrapped bacteria and NDPs9,10,24 causing symptoms analogous to the Jarisch-Herxheimer reaction.25
We propose an alternative strategy of NET-directed therapy in sepsis, in which NETs are compacted and stabilized, leading to both enhanced bacterial entrapment and NDP sequestration. Platelet factor 4 (PF4, CXCL4), a positively charged chemokine that accounts for ∼2% to 3% of total protein found in platelets,26 may exert these effects. When platelets are activated, PF4 is released in local concentrations that exceed 12 µg/mL.26 High levels of PF4 have been identified in the α-granules of every examined mammalian species, suggesting that it serves a crucial function, but its role remains unclear. Because of its strong positive charge, PF4 binds to and aggregates polyanions such as heparin.27 We have found that PF4 similarly aggregates NETs, compacting them and enhancing their resistance to endogenous and microbial nucleases.28,29 We speculated that these activities may significantly contribute to our previous observation that PF4-deficient mice (cxcl4−/−) have increased mortality in lipopolysaccharide (LPS) endotoxemia, whereas animals that overexpress human PF4 (hPF4+) have improved survival.30 These findings suggest that PF4’s protective role in sepsis may be the evolutionary force driving its conserved expression.
KKO is a monoclonal antibody directed against human (h) PF4-heparin complexes that induce thrombocytopenia and a prothrombotic state when injected into mice that express hPF4 and FcγRIIA, mimicking the clinical disorder heparin-induced thrombocytopenia (HIT).31 KKO also binds hPF4-NET complexes, further enhancing DNase resistance.29 We now speculate that treatment with an Fc-modified KKO that does not activate platelets via FcγRIIA or fix complement may augment the protective effect of PF4 in sepsis by stabilizing hPF4-NET complexes. To investigate this hypothesis, we generated an Fc-modified KKO that retains the ability to bind hPF4-NET complexes, but has a reduced capacity to stimulate the inflammatory response. In mice treated with LPS32 and in the murine cecal ligation and puncture (CLP) model of polymicrobial sepsis,32 treatment with this modified KKO led to protection from thrombocytopenia, decreased plasma NDP levels, reduced bacterial dissemination, and improved survival.
Methods
Antibodies studied and recombinant hPF4
KKO is a mouse immunoglobulin G2bκ (IgG2bκ) anti-hPF4/heparin monoclonal antibody.31 TRA is a monoclonal IgG isotype control antibody.31 Both KKO and TRA were purified from hybridoma supernatants.31 KKO and TRA were deglycosylated using EndoS (IgGZERO, Genovis)2 adding 1 U IgGZERO to 1 µg of KKO or TRA incubated at 37°C for 2 hours.33 Liquid chromatography with tandem mass spectrometry34 was used to confirm removal of the Fc glycan moieties from KKO (supplemental Figure 6, available on the Blood Web site). Recombinant hPF4 was expressed in S2 cells35 and purified using affinity chromatography and protein liquid chromatography as previously described.35 The end-product purity was found to be endotoxin free and was tested for size distribution by gel electrophoresis (sodium dodecyl sulfate-polyacrylamide gel electrophoresis). Deglycosylated (DG)-KKO’s ability to bind to PF4-heparin and yield an immunogenic product was confirmed by enzyme-linked immunosorbent assay (ELISA).35 Mouse plasma levels of murine PF4 and hPF4 were measured using commercially available ELISA kits (R&D Systems).
NET-lined microfluidic channel studies
NET-lined channels were generated as described previously.29 Human neutrophils were isolated as in the supplemental Methods. Cells (2 × 106 cells/mL) were incubated with 10 ng/mL tumor necrosis factor-α (TNF-α; Gibco) and allowed to adhere to channels coated with fibronectin (Sigma-Aldrich).36 Channels were then incubated with 100 ng/mL phorbol 12-myristate 13-acetate (Sigma-Aldrich) in Hanks buffered salt solution with calcium and magnesium (HBSS, Gibco) overnight at 37°C. cfDNA was visualized with 1 μM SYTOX green or orange (Thermo Fisher Scientific), also suspended in HBSS. Channels were then infused with hPF4 (0-100 μg/mL). Some channels containing PF4-NET complexes were incubated with 25 μg/mL of KKO, DG-KKO, or a polyclonal anti-PF4 antibody (25 µg/mL, Abcam) for 1 hour at 37°C. NET digestion studies were performed by infusing the channels with 100 U/mL of recombinant human DNase I (Sigma-Aldrich), as described elsewhere.29 KKO and DG-KKO were labeled with Alexa Fluor 647 (Thermo Fisher Scientific) before channel infusion. All infusions were done at 2 to 5 dynes/cm2. Experiments were performed using a BioFlux 200 Controller (Fluxion).37
Endothelialized channel microfluidic studies
Human umbilical vein endothelial cells (HUVECs, Lonza) at passage 3 to 4 (5 × 106 cells) were seeded onto fibronectin-coated (50 μg/mL, Sigma-Aldrich) channels of 48-well BioFlux plates (Fluxion) and cultured at 37°C under 5% CO2 in endothelial cell growth media (Lonza) for 2 to 3 days to become confluent. HUVECs were incubated with TNF-α (Gibco, 1 ng/mL) to simulate inflammation. Neutrophils were isolated from whole blood samples obtained from healthy human donors as described in the supplemental Methods, resuspended in HBSS, and treated with LPS (100 ng/mL) with and without PF4 (25 µg/mL) immediately before channel infusion at 2 dynes/cm2, where they adhered to the HUVECs and released NETs. Channels were also infused with Hoechst (Invitrogen, 1 µg/mL) to label all HUVEC nuclei. Flow was discontinued and channels were incubated at 37°C. HUVEC nuclei were counted at 16 hours. The BioFlux channels were visualized with an Axio Observer Z1 inverted Zeiss microscope equipped with a motorized stage and an HXP-120 C metal halide illumination source. The microscope and image acquisition were controlled by BioFlux Montage software with a MetaMorph-based platform (Molecular Devices). Data were analyzed using ImageJ open source image processing software.29
Alexa Fluor 488-labeled, heat-killed Staphylococcus aureus (Wood strain without lipid A) and Escherichia coli (K-12) bioparticles were used in the bacterial capture studies (6 × 106 bioparticles/mL, Molecular Probes). They were suspended in HBSS and infused for 30 minutes at 1 dynes/cm2 into NET channels previously treated with HBSS ± hPF4 at concentrations of 10, 25, and 100 μg/mL. Bacterial adhesion was quantified by a blinded observer using ImageJ to count adherent bioparticles. Some channels were then infused with DNase I (100 U/mL, Sigma-Aldrich) for 15 minutes. Other channels were infused with heparin (100 U/mL, Novaplus) for 30 minutes to remove PF4 from compacted NETs. The channels were then infused with Cellfix (BD Biosciences) for 15 minutes before imaging with a Zeiss LSM 710 laser-scanning confocal microscope.
Mice studied
Mice were wild-type (WT) C57Bl6 or littermates deficient in murine PF4 (cxcl4−/−) or transgenic for platelet-specific expressed hPF4 on a cxcl4−/− background (termed hPF4+ mice)38 because murine PF4 is not recognized by KKO and by most patient HIT antibodies.39 We have previously found that hPF4+ mice have approximately 2 times the amount of hPF4 per milligram of total platelet protein as human platelets, which is expressed during megakaryopoiesis.40 hPF4+ mice that express human FcγRIIA41 were used in a HIT passive immunization model35 to assess DG-KKO’s ability to induce thrombocytopenia. Genetic modifications were confirmed by polymerase chain reaction analyses.42 All mice were studied between 10 and 20 weeks of age. There was an equal distribution of gender in the mice generated, and animals of each gender were randomly studied.
LPS endotoxemia sepsis model
Baseline platelet counts from retro-orbital blood samples were measured using the Hemavet 950 (Drew Scientific). On the following day, WT, cxcl4−/−, or hPF4+ mice received an intraperitoneal (IP) injection of LPS (35 mg/kg). Thirty minutes later, the mice received a tail vein injection containing either vehicle alone or KKO, DG-KKO, TRA, or DG-TRA at doses of 5 mg/kg diluted in 200 μL of phosphate buffered saline (PBS with CaCl2 and MgCl2, Gibco). Six hours after LPS treatment, blood was collected from the retro-orbital plexus and platelet counts were measured. A subset of mice was treated with recombinant DNase I (20 mg/kg, IP, Sigma-Aldrich) 2 hours after LPS injection.24 Six hours following LPS injection, a subgroup of animals was euthanized, blood was collected from the inferior vena cava (IVC), and plasma was isolated. cfDNA was measured using a SYTOX fluorescent plate assay,43 MPO-cfDNA was measured using a previously described ELISA,44 and monocyte chemoattractant (MCP)-1 levels were quantified with western blot, using anti-MCP-1 (Cell Signaling Technology, #2027). Another subgroup was observed and the clinical murine sepsis score (MSS), a reproducible scoring system designed to evaluate the severity of illness in murine sepsis45 based on 7 clinical variables including general appearance, level of consciousness, activity, response to stimulus, eye opening, respiratory rate, and respiratory quality,45 was calculated and survival noted every 12 hours as described46 for up to 72 hours. When MSS exceeded 18, the animals were euthanized in accordance with Institutional Animal Care and Use Committee protocol.
CLP polymicrobial sepsis model
CLP-induced sepsis was induced as described47 in WT, cxcl4−/−, and hPF4+ mice. Mice were anesthetized using IP ketamine (150 mg/kg) and xylazine (10 mg/kg) before surgical exposure of the cecum.48 The cecum was exteriorized and ligated with a 6.0 silk suture (6-0 Proline, 8680G; Ethicon) placed below the ileo-cecal valve and perforated with a 21-gauge needle (BD Biosciences) to induce mid-grade lethal sepsis.49-51 After removing the needle, a small amount of feces was extruded. The cecum was relocated, and the fascia, abdominal musculature, and peritoneum were closed via a running suture. The control mice were anesthetized and underwent laparotomy without puncture or cecal ligation. Following the procedure, 1 mL of saline was administered subcutaneously,52 and the animals received a tail vein injection of PBS, KKO, DG-KKO, TRA, or DG-TRA at 5 mg/kg 30 minutes later. A subset of hPF4+ mice were treated with IP DNase I (20 mg/kg) 2 hours following surgery. In the WT mouse studies, the animals were given 20 or 40 mg/kg of hPF4 by tail vein injection immediately following surgery. All animals were treated with 0.05 mg/kg buprenorphine every 12 hours to maintain analgesia. In some experiments, ceftriaxone was injected intradermally immediately following surgery (100 mg/kg, Tocris Bioscience). In a subset of mice, 24 hours after surgery, blood was collected from the retro-orbital plexus and platelet counts were measured with the Hemavet 950FS. Another subset of animals was euthanized at 48 hours, and blood was obtained from the IVC to measure NDP levels. Peripheral blood colony forming units (CFUs) were measured by diluting 500 µL of whole blood in 5 mL of PBS, and plating 10 µL of the sample at 1:10, 1:100, and 1:1000 dilutions on agar plates. Liver CFUs were measured at the same dilutions after tissue was homogenized by pressing it through a 70-µm cell strainer (Fisher Scientific). Agar plates were incubated overnight in duplicate before individual colonies were counted. Another subgroup had MSS calculated and survival noted every 12 hours for up to 120 hours.
Statistical analysis
Differences between 2 groups were compared using a Mann-Whitney U test. Differences between more than 2 groups were determined with a Kruskal-Wallis 1-way analysis of variance (ANOVA) or with Sidak’s multiple comparisons tests, as appropriate. Multiplicity corrected P values are reported for multiple comparisons. Survival analysis was performed using a log-rank (Mantel-Cox) test. Statistical analyses were performed using GraphPad Prism 7.0 (GraphPad Software). Differences were considered statistically significant when P values were ≤ .05.
Study approval and additional methodology
Animal procedures were approved by the Institutional Animal Care and Use Committee at the Children’s Hospital of Philadelphia in accordance with National Institute of Health guidelines and the Animal Welfare Act. Anonymized human blood was collected after signed, informed consent was provided by healthy donors. Approval for the use of human blood was obtained from Children’s Hospital of Philadelphia’s Human Review Board in accordance with Declaration of Helsinki Principles.
Additional methodologies
The supplemental Methods contains a description of antibodies and other labeled probes, activated protein C (APC) generation assay, isolation of human neutrophils, and bioassays for NDPs.
Results
The effect of PF4 on plasma levels of NDPs in murine endotoxemia
We have previously observed that cxcl4−/− mice have increased mortality in LPS endotoxemia compared with hPF4+ animals that overexpress hPF4, and that infusion of WT mice with hPF4+ platelets improved survival.30 We have recently found that PF4 binds to NETs, leading them to become physically compact and DNase resistant.29 Using an in vitro microfluidic system,29 we now show that mouse PF4 (mPF4) also compacts NETs and protects them from DNase digestion (supplemental Figure 1). Therefore, we chose to examine whether NET compaction with NDP sequestration contribute to the protective effects of PF4 in sepsis. We measured NDP levels, including cfDNA, MPO, histone H3, and citrullinated histone H3 (cit-H3) in cxcl4−/− and hPF4+ mice following LPS injection. cxcl4−/− mice had higher levels of cfDNA and MPO than hPF4+ animals after LPS exposure (Figure 1A-B). H3 levels were on average higher in cxcl4−/− mice following LPS injection, but this difference was not significant. In comparison, levels of cit-H3, a more specific marker of NETosis,53 were significantly elevated in cxcl4−/− animals (Figure 1C). We next explored whether infused mPF4 could recapitulate the protective effect of endogenous PF4 when administered via osmotic pumps. We also examined whether hPF4 may have a similar effect when administered as a single IV bolus. Eight hours following osmotic pump placement and tail vein injections, plasma mPF4 and hPF4 levels rose significantly (supplemental Figure 2). We measured plasma levels of cfDNA and MPO-cfDNA complexes 6 hours after LPS treatment and found both approaches led to decreased plasma NDPs (Figure 1D-F).
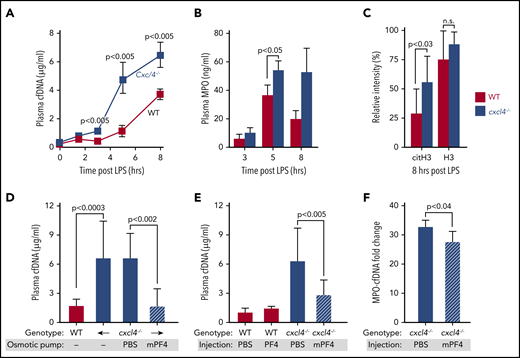
Effects of murine and human PF4 on circulating NDP levels following LPS exposure in WT and cxcl4−/−mice. (A) WT and cxcl4−/− mice received LPS (35 mg/kg, IP). Plasma samples were obtained at the indicated time points. Mean plasma cfDNA levels are shown as ± 1 standard deviation (SD). N = 7-14 mice per arm. P values are indicated comparing WT and cxcl4−/− mice using a Mann-Whitney U test. (B) The same as panel A, except for MPO levels at 3 to 8 hours post-LPS. (C) Histone levels comparing western blot intensity to that of the positive control band. N = 9-10 mice per arm. P values are indicated comparing WT and cxcl4−/− mice by Mann-Whitney U test. (D) LPS studies comparing WT and cxcl4−/− mice following implantation of osmotic pumps containing PBS alone or 80 µg of mPF4. Mean ± 1 SD are shown N = 5-8 mice per arm. (E) The same as panel D, except following a single dose of hPF4 (330 µg, IV). N = 5-8 mice per arm. (F) LPS studies comparing plasma levels of MPO-cfDNA complex in cxcl4−/− mice that received tail vein injections containing PBS alone or 330 µg of hPF4. N = 4-5 mice per arm. P values are indicated comparing WT and cxcl4−/− mice by Mann-Whitney U test.
Effects of murine and human PF4 on circulating NDP levels following LPS exposure in WT and cxcl4−/−mice. (A) WT and cxcl4−/− mice received LPS (35 mg/kg, IP). Plasma samples were obtained at the indicated time points. Mean plasma cfDNA levels are shown as ± 1 standard deviation (SD). N = 7-14 mice per arm. P values are indicated comparing WT and cxcl4−/− mice using a Mann-Whitney U test. (B) The same as panel A, except for MPO levels at 3 to 8 hours post-LPS. (C) Histone levels comparing western blot intensity to that of the positive control band. N = 9-10 mice per arm. P values are indicated comparing WT and cxcl4−/− mice by Mann-Whitney U test. (D) LPS studies comparing WT and cxcl4−/− mice following implantation of osmotic pumps containing PBS alone or 80 µg of mPF4. Mean ± 1 SD are shown N = 5-8 mice per arm. (E) The same as panel D, except following a single dose of hPF4 (330 µg, IV). N = 5-8 mice per arm. (F) LPS studies comparing plasma levels of MPO-cfDNA complex in cxcl4−/− mice that received tail vein injections containing PBS alone or 330 µg of hPF4. N = 4-5 mice per arm. P values are indicated comparing WT and cxcl4−/− mice by Mann-Whitney U test.
In vitro protective effects of PF4
In sepsis, end-organ injury involves damage to the microvasculature.54 Therefore, we sought to determine if PF4-mediated compaction alters NETs’ ability to harm the endothelium. To that end, we stimulated isolated human neutrophils with LPS and resuspended the cells in media with and without hPF4 and flowed the samples through HUVEC-lined microfluidic channels that had been stimulated with TNF-α. The neutrophils readily adhered to the endothelial monolayer, releasing NETs that were fluorescently labeled with the live cell membrane impermeant nucleic acid stain, SYTOX green. In prior studies, these structures were also labeled with MPO and cit-H3 to confirm their identity as NETs.29 The presence of PF4 led to NET compaction, although the cfDNA continued to fill the width of the channels (supplemental Figure 3). Flow was discontinued and the HUVECs were incubated with the NET-containing media for 16 hours, after which channels treated with PF4 contained a higher number of residual adherent endothelial cells, suggesting that PF4-bound NETs are less harmful to the endothelium (Figure 2A).
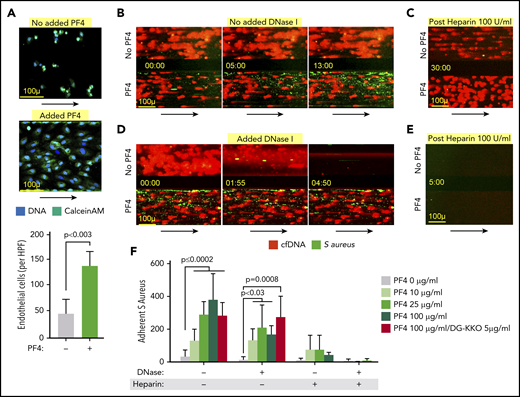
Effect of PF4 on endothelial cells and microbial entrapment by NETs in vitro. Channels lined with TNF-α–stimulated HUVECs were infused with isolated human neutrophils treated with LPS (100 ng/mL) to induce NETosis with and without hPF4 (25 µg/mL). (A) Channels were incubated with the neutrophils for 16 hours, after which the number of residual adherent endothelial cells was counted using ImageJ. (Top) Representative images of remaining attached endothelial cells channels per condition. Size bar and arrows indicate direction of flow. Bottom: mean of endothelial cell counts in 3 ×10 high-powered fields per condition ± 1 SD. Statistical analysis was performed using a Mann-Whitney U test. n = 6 channels per arm. (B) Left shows representative images of NET-lined channels infused with fluorescently labeled S aureus with observed bacterial capture. Size bar and arrows indicating direction of flow are included. NETs in bottom channels compacted with PF4 (100 μg/mL). (C) Representative image of NET-lined channels previously infused with bacteria, following 30-minute infusion of heparin 100 U/mL. (D) The same as panel C, but includes images of NET-lined channels infused with S aureus bioparticles followed by a 5-minute infusion of DNase I (100 U/mL). NETs in bottom channels compacted with PF4 (100 μg/mL). (E) The same as panel C showing image of S aureus–infused channels previously treated with heparin, now following infusion with DNase 1 (100 U/mL). (F) Graph showing number of NET-adherent bacterial bioparticles in channels ± 1 SD following the initial infusion, DNase I (100 U/mL) × 15 minutes, or heparin (100 U/mL) × 30 minutes as indicated. N = 3-15 channels per condition. Analysis performed by a Kruskal-Wallis 1-way ANOVA.
Effect of PF4 on endothelial cells and microbial entrapment by NETs in vitro. Channels lined with TNF-α–stimulated HUVECs were infused with isolated human neutrophils treated with LPS (100 ng/mL) to induce NETosis with and without hPF4 (25 µg/mL). (A) Channels were incubated with the neutrophils for 16 hours, after which the number of residual adherent endothelial cells was counted using ImageJ. (Top) Representative images of remaining attached endothelial cells channels per condition. Size bar and arrows indicate direction of flow. Bottom: mean of endothelial cell counts in 3 ×10 high-powered fields per condition ± 1 SD. Statistical analysis was performed using a Mann-Whitney U test. n = 6 channels per arm. (B) Left shows representative images of NET-lined channels infused with fluorescently labeled S aureus with observed bacterial capture. Size bar and arrows indicating direction of flow are included. NETs in bottom channels compacted with PF4 (100 μg/mL). (C) Representative image of NET-lined channels previously infused with bacteria, following 30-minute infusion of heparin 100 U/mL. (D) The same as panel C, but includes images of NET-lined channels infused with S aureus bioparticles followed by a 5-minute infusion of DNase I (100 U/mL). NETs in bottom channels compacted with PF4 (100 μg/mL). (E) The same as panel C showing image of S aureus–infused channels previously treated with heparin, now following infusion with DNase 1 (100 U/mL). (F) Graph showing number of NET-adherent bacterial bioparticles in channels ± 1 SD following the initial infusion, DNase I (100 U/mL) × 15 minutes, or heparin (100 U/mL) × 30 minutes as indicated. N = 3-15 channels per condition. Analysis performed by a Kruskal-Wallis 1-way ANOVA.
We then investigated how PF4 binding influences NETs’ ability to trap bacteria, another important way NETs affect outcome in sepsis.9 We infused NET-lined channels with either heat-inactivated, fluorescently labeled S aureus or E coli bioparticles, and quantified bacterial capture by wide-field microscopy. PF4-compacted NETs captured a higher number of bacteria (Figure 2B; supplemental Figure 4; supplemental Videos 1 and 2) that adhered directly to the NET fibers (supplemental Figure 5). Although we observed this effect at physiologic concentrations of PF4 (10 μg/mL), it became more pronounced at supraphysiologic concentrations of 25 and 100 μg/mL (Figure 2D). Moreover, when these channels were infused with recombinant human DNase I, PF4-free NETs were rapidly digested, liberating captured bacteria (Figure 2B, bottom; supplemental Video 3). In contrast, during DNase I infusion, compacted PF4-bound NETs remained intact and did not release immobilized bacteria (Figure 2D; supplemental Video 3). There was no significant difference between the numbers of adherent bacteria in NETs compacted with PF4 10 to 25 μg/mL with or without DNase I administration (Figure 2F). Although there was a statistically significant reduction in adherent bacteria in NETs compacted with PF4 100 μg/mL following DNase I infusion (P < .0007, analysis with Mann-Whitney U test), the number of residual adherent bacteria in these channels was significantly higher than that seen in NETs that were not treated with PF4 (P < .01, analysis with Mann-Whitney U test). This decrease in entrapment was not observed in NETs treated with PF4 100 μg/mL + DG-KKO, in which the numbers of adherent bacteria remained stable after DNase I infusion (Figure 2F). To confirm that the increase in bacterial adhesion was because of PF4, we infused the channels with high doses of heparin (100 U/mL), which removes PF4 from NETs, leading to NET decompaction.29 We observed that the removal of PF4 with heparin did not induce NET lysis, but did lead to the release of bacteria from NETs (Figure 2C,F; supplemental Video 4). Following PF4 removal, decompacted NETs again became susceptible to lysis with DNase I, which now induced the release of nearly all remaining bacteria (Figure 2E-F).
Studies of the in vitro effects of DG-KKO
By the time most patients present with symptoms of sepsis, their platelets have released large amounts of PF455 that can avidly bind circulating cfDNA.28 Our prior observation that the HIT-like monoclonal antibody KKO enhances DNase resistance of hPF4-NET complexes29 raised the possibility that KKO may amplify the effect of endogenous PF4 and be of therapeutic benefit in sepsis. However, KKO fixes complement,56 activates platelets,31 and stimulates leukocytes,29,57 inducing a prothrombotic state.36 We modified KKO by deglycosylating the antibody58 to decrease Fc-mediated injury. DG-KKO was >97% deglycosylated by liquid chromatography with tandem mass spectrometry34 (supplemental Figure 6). DG-KKO continued to bind hPF4-heparin complexes and hPF4-glycosaminoglycan complexes on the platelet surface (Figure 3A-B), but had a reduced capacity to activate hPF4-exposed human platelets or induce thrombocytopenia in hPF4+/FcγRIIA+ mice (Figure 3C-D). In studies with NET-lined microfluidic channels, DG-KKO did not adhere to noncompacted NETs, but bound hPF4-compacted NETs (Figure 3E). As with KKO, DG-KKO also increased hPF4-NET resistance to DNase I under flow conditions in microfluidic channels (Figure 3F-G; supplemental Video 5) without impacting the ability of PF4-NET complexes to capture bacteria (supplemental Figure 5; supplemental Video 6).
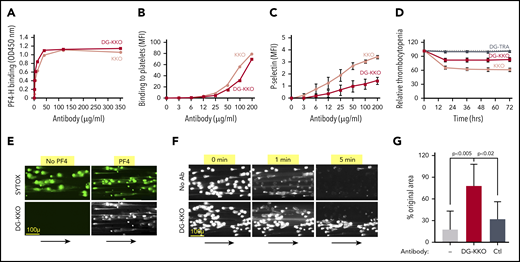
Binding of DG-KKO to PF4/NET complexes in vitro. (A) Graphs quantifying binding of increasing concentrations of KKO (gray) and DG-KKO (red) to PF4-heparin (PF4-H) using fluorescent plate assay. (B) The same as panel A but using flow cytometry to quantify antibody binding to the platelet surface. (C) Mean ± 1 SD of P-selectin MFI in human whole blood samples incubated with the indicated concentration of antibody, reflecting the degree of platelet activation. N = 3. (D) Mean ± 1 SD of the % decrease in platelets counts in HIT mice injected with 400 µg of the indicated antibody, measured every 12 hours for 3 days. N = 10. (E) Representative confocal images of released NETs as in Figure 2 exposed to no PF4 or 6.5 µg/mL of PF4, labeled with the nucleic acid stain SYOTX green (green) demonstrating change in morphology. The indicated channels were then infused with fluorophore-labeled DG-KKO (white). Size bar and arrows indicating direction of flow are included. Image were obtained at ×10 magnification. (F) Representative widefield images of adherent NETs as in panel A, but in the presence of 100 U/mL DNase I and 6.5 µg/mL PF4 ± 25 µg/mL of DG-KKO. (G) Mean ± 1 SD of the relative area of NETs compacted with PF4 (6.5 µg/mL) alone or with PF4 plus DG-KKO or a polyclonal anti-PF4 antibody control (Ctl) (each, 25 µg/mL) after an infusion of DNase I (100 U/mL, 3 minutes) compared with preinfusion area. N = 7-10 channels per condition. Comparative statistical analysis performed by Kruskal-Wallis 1-way ANOVA.
Binding of DG-KKO to PF4/NET complexes in vitro. (A) Graphs quantifying binding of increasing concentrations of KKO (gray) and DG-KKO (red) to PF4-heparin (PF4-H) using fluorescent plate assay. (B) The same as panel A but using flow cytometry to quantify antibody binding to the platelet surface. (C) Mean ± 1 SD of P-selectin MFI in human whole blood samples incubated with the indicated concentration of antibody, reflecting the degree of platelet activation. N = 3. (D) Mean ± 1 SD of the % decrease in platelets counts in HIT mice injected with 400 µg of the indicated antibody, measured every 12 hours for 3 days. N = 10. (E) Representative confocal images of released NETs as in Figure 2 exposed to no PF4 or 6.5 µg/mL of PF4, labeled with the nucleic acid stain SYOTX green (green) demonstrating change in morphology. The indicated channels were then infused with fluorophore-labeled DG-KKO (white). Size bar and arrows indicating direction of flow are included. Image were obtained at ×10 magnification. (F) Representative widefield images of adherent NETs as in panel A, but in the presence of 100 U/mL DNase I and 6.5 µg/mL PF4 ± 25 µg/mL of DG-KKO. (G) Mean ± 1 SD of the relative area of NETs compacted with PF4 (6.5 µg/mL) alone or with PF4 plus DG-KKO or a polyclonal anti-PF4 antibody control (Ctl) (each, 25 µg/mL) after an infusion of DNase I (100 U/mL, 3 minutes) compared with preinfusion area. N = 7-10 channels per condition. Comparative statistical analysis performed by Kruskal-Wallis 1-way ANOVA.
In previous studies, we have shown that treatment with PF4 leads to accelerated APC generation, which ameliorates coagulopathy and endothelial damage in sepsis.30 We therefore speculated that DG-KKO may influence the rate of APC generation by binding to PF4 complexed with polyanions. To determine if this may be the case, we used a previously described in vitro APC generation assay59 combining soluble thrombomodulin, protein C, and factor IIa with hPF4 alone or in combination with KKO or DG-KKO. We observed that the addition of either KKO or DG-KKO led to a near-identical decrease in the rate of APC generation (supplemental Figure 7). Thus, if DG-KKO is beneficial in murine sepsis models, this effect is not due to increased APC generation.
DG-KKO reduces plasma NDPs and improves outcomes in murine LPS endotoxemia
We next investigated whether treatment with DG-KKO improves outcomes in murine LPS endotoxemia. Thirty minutes after LPS exposure, mice were given either DG-KKO or a deglycosylated version of an isotype control antibody, DG-TRA,35 via tail-vein injection. Because both KKO and DG-KKO bind specifically to NET complexes containing hPF4, and not complexes with mPF4 (Figure 3E and Li et al39 ), hPF4+ mice were studied. We observed that LPS-exposed hPF4+ mice treated with DG-KKO were protected from thrombocytopenia (Figure 4A). Moreover, animals treated with DG-KKO had lower plasma levels of cfDNA and MPO-cfDNA complexes (Figure 4B-C) and a decrease in the inflammatory cytokine MCP-1 (Figure 4D). DG-KKO–treated animals did not have elevated plasma levels of thrombin antithrombin complexes (supplemental Figure 8), indicating that DG-KKO stabilization of NETs did not accelerate thrombin generation.60 DG-KKO–treated mice also had a more benign clinical course, which was demonstrated by significantly lower mean clinical MSS at 12 hours (Figure 4E) and improved overall survival (Figure 4F). In parallel LPS studies of WT and cxcl4−/− mice, DG-KKO did not exert a protective effect (Figure 4A-F), supporting that DG-KKO acts through an hPF4-dependent pathway.
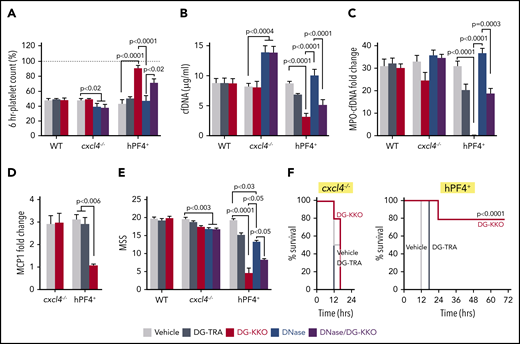
DG-KKO treatment in WT, cxcl4−/−, and hPF4+mice undergoing LPS endotoxemia. Mice were injection with LPS (35 mg/kg, IP). Thirty minutes later, they received tail vein injections containing vehicle alone or 5 mg/kg of DG-KKO or DG-TRA isotype control or DG-KKO plus DNase I (20 mg/kg). Six hours following LPS injection, a subset of mice was euthanized and IVC blood samples were collected. (A) Relative to baseline (dashed gray line), 6-hour time point platelet counts. Mean ± 1 SD shown. (B-E) The same as panel A, but for cfDNA concentration, MPO-cfDNA complex fold change relative to negative control, MCP-1 levels as measured by fold change in western blot bandwidth relative to GAPDH control, and MSS. N = 3-10 in each arm. Comparative statistical analysis was performed with a Kruskal-Wallis 1-way ANOVA. (F) Animal survival results for the LPS studies in the cxcl4−/− mice and hPF4+ mice. Results were analyzed with the log-rank test. n = 5 animals per arm.
DG-KKO treatment in WT, cxcl4−/−, and hPF4+mice undergoing LPS endotoxemia. Mice were injection with LPS (35 mg/kg, IP). Thirty minutes later, they received tail vein injections containing vehicle alone or 5 mg/kg of DG-KKO or DG-TRA isotype control or DG-KKO plus DNase I (20 mg/kg). Six hours following LPS injection, a subset of mice was euthanized and IVC blood samples were collected. (A) Relative to baseline (dashed gray line), 6-hour time point platelet counts. Mean ± 1 SD shown. (B-E) The same as panel A, but for cfDNA concentration, MPO-cfDNA complex fold change relative to negative control, MCP-1 levels as measured by fold change in western blot bandwidth relative to GAPDH control, and MSS. N = 3-10 in each arm. Comparative statistical analysis was performed with a Kruskal-Wallis 1-way ANOVA. (F) Animal survival results for the LPS studies in the cxcl4−/− mice and hPF4+ mice. Results were analyzed with the log-rank test. n = 5 animals per arm.
Early administration of DNase I is less protective than DG-KKO in LPS endotoxemia
We compared the effects of DG-KKO to treatment with DNase in parallel LPS studies. We observed that nuclease treatment led to a significant rise in plasma cfDNA levels in cxcl4−/− mice without a significant effect in hPF4+ mice, suggesting that NETs in cxcl4−/− animals may be more vulnerable to nuclease cleavage (Figure 4B). Although hPF4+ animals treated with a combination of DNase I and DG-KKO had decreased plasma concentrations of cfDNA and MPO-cfDNA complexes compared with those treated with DNase I alone, they continued to have lower platelet counts and greater plasma NDP levels than animals treated exclusively with DG-KKO, showing that DG-KKO could only partially rescue animals from the effect of DNase I (Figure 4B-D). In both cxcl4−/− and hPF4+ mice, treatment with DNase I did lead to a significant reduction in MSS. However, the protective effect of DNase I alone and in combination with DG-KKO was less robust than that seen with DG-KKO monotherapy (Figure 5E), supporting our contention that NET compaction rather than enhanced NET cleavage is the superior treatment modality in LPS endotoxemia.
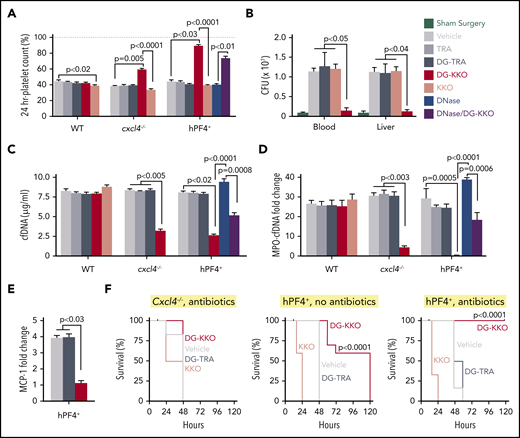
DG-KKO treatment in cxcl4−/−and hPF4+mice undergoing CLP injury. All the mice underwent CLP procedure. Immediately following surgery, a subset of mice received an intradermal dose of the antibiotic ceftriaxone (100 mg/kg). Mice also were divided by therapeutic intervention, receiving either vehicle only or 5 mg/kg of TRA, DG-TRA, KKO, or DG-KKO. After 24 hours, platelet counts were quantified. After 48 hours, bacterial CFUs, plasma NDP levels, and survival were measured. Mean ± 1 SD shown. (A) Relative platelet counts 24 hours after CLP injury measured as in Figure 4A. (B) CFUs as measured in the peripheral blood and liver homogenates of animals 48 hours following CLP. Black bars indicate animals that underwent sham surgery. (C-D) The same as Figure 4B-C, respectively, but 48 hours after CLP injury. (A-D) N = 6-10 animals per arm. Statistical analysis performed with a Kruskal-Wallis 1-way ANOVA. (E) Plasma MCP-1 levels in hPF4+ mice, measured as in Figure 4D. n = 5. Analysis with a Kruskal-Wallis 1-way ANOVA. (F) Animal survival results for the CLP studies. N = 10. Statistical analysis performed with a log-rank (Mantel-Cox) test, showing a significant increase in survival in hPF4+ mice treated with DG-KKO compared with those treated with vehicle alone, KKO, or DG-TRA with and without antibiotics (P < .0001).
DG-KKO treatment in cxcl4−/−and hPF4+mice undergoing CLP injury. All the mice underwent CLP procedure. Immediately following surgery, a subset of mice received an intradermal dose of the antibiotic ceftriaxone (100 mg/kg). Mice also were divided by therapeutic intervention, receiving either vehicle only or 5 mg/kg of TRA, DG-TRA, KKO, or DG-KKO. After 24 hours, platelet counts were quantified. After 48 hours, bacterial CFUs, plasma NDP levels, and survival were measured. Mean ± 1 SD shown. (A) Relative platelet counts 24 hours after CLP injury measured as in Figure 4A. (B) CFUs as measured in the peripheral blood and liver homogenates of animals 48 hours following CLP. Black bars indicate animals that underwent sham surgery. (C-D) The same as Figure 4B-C, respectively, but 48 hours after CLP injury. (A-D) N = 6-10 animals per arm. Statistical analysis performed with a Kruskal-Wallis 1-way ANOVA. (E) Plasma MCP-1 levels in hPF4+ mice, measured as in Figure 4D. n = 5. Analysis with a Kruskal-Wallis 1-way ANOVA. (F) Animal survival results for the CLP studies. N = 10. Statistical analysis performed with a log-rank (Mantel-Cox) test, showing a significant increase in survival in hPF4+ mice treated with DG-KKO compared with those treated with vehicle alone, KKO, or DG-TRA with and without antibiotics (P < .0001).
DG-KKO improves outcomes in murine CLP polymicrobial sepsis model
Although the LPS endotoxemia model recapitulates many aspects of sepsis, it reflects the effect of a bacterial toxin rather than live pathogens. To determine if PF4 and DG-KKO mitigate NET-associated toxicities when bacteria are present, we repeated our endotoxemia experiments using CLP to induce polymicrobial sepsis.61 We performed these experiments in cxcl4−/− and hPF4+ mice with and without the administration of the antibiotic ceftriaxone given at a dose that does not improve outcome when used as a single agent.62 We observed no protective effect of DG-KKO in cxcl4−/− mice following CLP (Figure 5). In contrast, DG-KKO prevented the development of thrombocytopenia in hPF4+ mice (Figure 5A). These animals also had a reduction in plasma cfDNA and MPO-cfDNA complex levels (Figure 5C-D), decreased levels of MCP-1 (Figure 5E), and prolongation of survival (Figure 5F). Animals cotreated with ceftriaxone and DG-KKO survived CLP (Figure 5F), demonstrating that NDP sequestration may complement conventional antibiotic strategies in the treatment of polymicrobial sepsis. The administration of DNase I to hPF4+ mice cotreated with ceftriaxone led to a significant increase in plasma levels of cfDNA and MPO-cfDNA complexes compared with that seen in animals treated with ceftriaxone alone (P = .0032 and P = .0009, respectively; n = 5-11, analysis with Mann-Whitney U test), similar to the result we observed in the LPS endotoxemia model of sepsis. Although treatment with DG-KKO before DNase I mitigated this effect, animals treated exclusively with DG-KKO had the lowest NDP levels (Figure 5C-D).
When bacterial CFUs were measured in the peripheral blood and livers 48 hours after CLP in ceftriaxone-treated hPF4+ mice, we found that DG-KKO infusion led to a significant reduction in blood and liver bacterial levels vs those seen in animals treated with sham surgery. Animals treated with DG-TRA and KKO had CFU levels similar to those treated with vehicle alone (Figure 5B).
Although we anticipated that KKO would have no effect in WT animals that do not express hPF4, we did observe that it led to a small but significant worsening of thrombocytopenia (Figure 5A) along with a slight but nonsignificant increase in mean plasma concentrations of cfDNA and MPO-cfDNA complexes. In contrast, hPF4+ mice treated with KKO had accelerated death compared with animals that received DG-TRA or vehicle alone (Figure 5F; P < .0006 in animals treated with ceftriaxone and P < .0001 in mice that did not receive antibiotics. n = 5 animals in each arm, analysis with log-rank test), and we were therefore unable to measure plasma levels of NDPs in these animals. This rapid demise of hPF4+ animals treated with KKO lends support to our thesis that Fc modification converts KKO from a pathogenic to a therapeutic agent in sepsis.
Coadministration of DG-KKO and hPF4 led to improved outcomes in CLP in WT mice
We next investigated whether DG-KKO is protective when coadministered with exogenous hPF4 in WT mice. These studies were designed to further support our proposed model and also define the exogenous dose of hPF4 needed to provide clinical benefit in conjunction with an Fc-modified KKO. We observed that WT mice treated with hPF4 alone at doses of 20 mg/kg were not significantly protected following CLP. However, treatment with 40 mg/kg led to protection from thrombocytopenia (Figure 6A) and lower plasma levels of cfDNA and MPO-cfDNA (Figure 6B-C). No protective effect was observed when animals were infused with DG-KKO and bovine serum albumin (data not shown). However, mice treated with a combination of DG-KKO and the lower dose of hPF4 (20 mg/kg) did well with the highest platelet counts (Figure 6A), the lowest plasma NDP levels (Figure 6B-C), and improved MSSs (Figure 6D). These results indicate it may be feasible to replicate our murine studies in models using larger animals that do not express hPF4 using a combination of exogenous hPF4 and DG-KKO to achieve maximal effect. These results also suggest that coinfusion of hPF4 with DG-KKO may be a superior therapeutic option to either treatment alone.
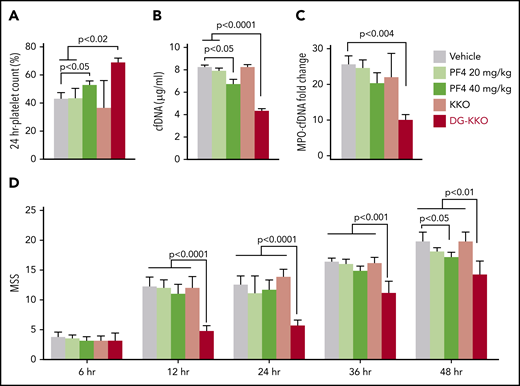
Treatment with hPF4 or hPF4 plus KKO or DG-KKO in WT mice undergoing CLP injury. CLP injuries were done as in Figure 5, but with WT mice that either received vehicle, hPF4 at either 20 or 40 mg/kg, or combination therapy of hPF4 (20 mg/kg) plus either 5 mg/kg KKO or DG-KKO. N = 10 animals per arm. (A) Relative platelet counts as in Figure 5A. (B-C) The same as in Figure 5C-D, respectively. (D) MSS in these studies for up to 48 hours after the injury. Mean ± 1 SD shown. (A-D) Statistical analysis was performed with Sidak’s multiple comparison t test.
Treatment with hPF4 or hPF4 plus KKO or DG-KKO in WT mice undergoing CLP injury. CLP injuries were done as in Figure 5, but with WT mice that either received vehicle, hPF4 at either 20 or 40 mg/kg, or combination therapy of hPF4 (20 mg/kg) plus either 5 mg/kg KKO or DG-KKO. N = 10 animals per arm. (A) Relative platelet counts as in Figure 5A. (B-C) The same as in Figure 5C-D, respectively. (D) MSS in these studies for up to 48 hours after the injury. Mean ± 1 SD shown. (A-D) Statistical analysis was performed with Sidak’s multiple comparison t test.
Discussion
Our group developed KKO in the early 2000s as a murine antibody directed against complexes of hPF4 and heparin, designed to mimic the pathogenic antibodies that induce HIT, a disorder characterized by a mild thrombocytopenia and a severe prothrombotic state.63 When injected into mice that express FcγRIIA and hPF4, KKO recapitulates this phenotype.64 Although HIT is classically thought of as a disorder of platelet activation, using this model, we and others observed that HIT antibodies bind to and activate neutrophils, causing them to release NETs that contribute to the development of thrombosis.29,65,66 In studies designed to investigate the role of neutrophils in HIT, we recently observed that PF4 binds to NETs, causing them to become physically compact and resistant to DNase I without displacing NDPs such as MPO and histones. We considered whether treatment with exogenous PF4 might improve outcomes in sepsis. However, by the time patients become clinically ill, most are likely to have a high level of platelet activation and PF4 release.4,67 Because platelet counts and PF4 levels per platelet vary greatly in the general population,68,69 we believe that some patients would have low circulating PF4 levels and would benefit from supplemental PF4 infusions, whereas others with sufficiently high platelet counts and platelet PF4 levels would receive little to no benefit. Therefore, an alternative, perhaps more effective treatment strategy would be an intervention that further stabilized interactions between NETs and endogenous PF4.
It had been shown that PF4 binding to DNA aptamers exposes HIT antigenic sites.70 Therefore, it was not surprising that the HIT-like monoclonal antibody KKO bound specifically to PF4-NET complexes, further enhancing their DNase resistance.29 However, KKO is an IgG2bκ antibody that activates Fc receptors and induces complement-dependent cytotoxicity after binding to complexes of hPF4 and polyanions.71 Therefore, unmodified KKO is injurious in sepsis, as shown in our studies of hPF4+ mice (Figure 5F). Our data showing that KKO slightly worsens thrombocytopenia and increases plasma NDPs following CLP in WT mice suggest that it may also be exerting harmful effects through PF4-independent mechanisms (Figure 5A,C-D). IgG2bκ antibodies are resistant to cleavage to form F(ab)2 fragments,72 and in studies not shown, we were unable to cleave KKO to generate such fragments. We therefore treated KKO with EndoS to generate deglycosylated DG-KKO.73 We speculate that KKO blocks DNase digestion of hPF4-NETs sterically so that DG-KKO may be a more efficacious product than a smaller F(ab)2 KKO fragment.
Other groups have used antibody deglycosylation in the treatment of immune-mediated disease. There is evidence that infusion with IdeS, a bacterial protease that cleaves the hinge region of heavy chain IgG, abolishing its ability to bind to FcγR, reduces the pathogenicity of antibodies in HIT, immune thrombocytopenia, and solid organ transplant.74-76 EndoS has also been used to deglycosylate pathogenic anti-aquaporin-4 antibodies in neuromyelitis optica, converting them into therapeutic blocking antibodies.77 Accordingly, we found that DG-KKO had a decreased ability to activate FcγRIIA. However, it retained the ability to stabilize PF4-NET complexes, decreasing NDP release and protecting the endothelial lining without interfering with NET bacterial entrapment. In murine models of sepsis, this translated to an hPF4-dependent decrease in thrombocytopenia, bacterial dissemination, cytokine levels, and NDP release, associated with improved survival.
Prior studies of NET-directed therapies in sepsis have focused on the blockade of NETosis through peptidyl arginine deiminase 4 (PAD4) inhibition or NET digestion with DNase. Our experiments with DNase I infusions are consistent with the results of prior murine CLP studies in which the early administration of DNase leads to increased inflammation and decreased survival.24 We similarly found that there was a greater increase in plasma NDP levels following DNase infusion in cxcl4−/− mice compared with hPF4+ mice, showing that PF4 protects NETs from DNase cleavage in vivo. However, even in hPF4+ mice, DNase I treatment led to elevation in plasma NDPs, suggesting that hPF4 does not fully abrogate this effect. Interestingly, when DG-KKO is administered following DNase I in hPF4+ mice, it continues to significantly reduce plasma NDP levels and MSS, suggesting that DG-KKO may remain effective even in the setting of the microbial release of nucleases.
The other NET-based therapeutic strategy that has been studied is the blockade of NETosis through PAD4 inhibition. Although PAD4 blockade may prove to be a useful treatment of autoimmune disease, it is an imperfect strategy in the treatment of sepsis as NETs entrap bacteria, which may enhance their killing.78 Preventing NETosis could lead to increased bacterial dissemination even in an era when effective antibiotics are often available. To date, published results investigating PAD4 inhibition in murine models of sepsis have been mixed, with some studies showing minimal negative effects21 and others revealing increased vulnerability to infection.9,79 The second problem with blocking NETosis is that by the time patients present with sepsis, their neutrophils have likely already released a large burden of NETs. In clinical studies of patients with septic shock, levels of MPO-cfDNA were found to be highest at the time of admission.19 Therefore, even prompt administration of a PAD4 inhibitor is likely to be ineffective in the treatment of most patients with sepsis.
Our results with PF4 and DG-KKO infusions suggest that in sepsis, NET compaction/stabilization, and NDP and microbial sequestration may be superior to PAD4 blockade or DNase infusion. We have found that it is protective in both sterile LPS endotoxemia and CLP polymicrobial sepsis. These results indicate that enhanced capture of bacteria by PF4 complexed to NETs dramatically improves the body’s ability to combat bacterial infections. However, in the setting of severe infection, as modeled by our CLP experiment, it is only when given with antibiotics that the animals consistently recover. These results suggest that NET-directed therapies are most effective when coadministered with antibiotics, and it is possible that these 2 treatment modalities may act synergistically to lead to optimal outcomes. Whether DG-KKO may also be effective in other conditions characterized by NET release including disseminated intravascular coagulation,80 sickle cell disease,81 and antiphospholipid antibody syndrome82 is unclear and needs to be tested in comparison with alternative strategies.
Finally, multiple functions have been attributed to PF4,83 but the reason for its conserved expression in all examined mammalian species remains unclear. We believe that insights from past studies of HIT and these studies of sepsis suggest that PF4’s biology is related to its ability to cross-aggregate multiple different polyanions, especially those released by neutrophils and NETs. We now show that PF4 limits NDP release, protects the endothelium, and enhances the ability of NETs to entrap both gram-positive and gran-negative organisms, providing survival advantages during LPS endotoxemia and polymicrobial infection. The strong evolutionary drive to protect against microbial invasion may explain the high concentration of this unusual chemokine in megakaryocytes and platelets.
For original data, please contact the corresponding author.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
This work was supported by the National Institutes of Health National Heart Lung and Blood Institute grants R01 HL139448 (M.P.) and T32HL007150 (K.G.), an American Heart Association Transformational Project Award (M.P.), and an American Society of Hematology Fellow Research Award (K.G.).
Authorship
Contribution: K.G. and A.S. carried out and evaluated these studies and prepared the first draft and subsequent revisions; M.A.K. conducted aPC and murine endotoxemia experiments and provided research guidance; L.R. helped to develop DG-KKO and provided research guidance; S.H. performed the Escherichia coli bacterial capture studies; S.H.S. performed the mass spectroscopy studies; G.M.A. developed KKO, provided research guidance, and edited the manuscript; K.H. provided guidance related to the measurement of bacterial dissemination and manuscript preparation; and M.P. provided overall project organization and direction, data interpretation, and manuscript preparation.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Mortimer Poncz, Children’s Hospital of Philadelphia, 3615 Civic Center Blvd, ARC, Rm 317, Philadelphia, PA 19104; e-mail: poncz@email.chop.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal