

In this issue of Blood, demonstrate that a monoclonal antibody (KKO) with properties resembling those of antibodies from patients with heparin-induced thrombocytopenia (HIT) can be deglycosylated and used therapeutically to improve outcomes in a murine model of sepsis.1 The mechanism underlying this activity appears to involve contraction and stabilization of neutrophil extracellular traps (NETs) and suggests a novel approach to the management of sepsis, which is associated with a mortality of 30% and an annual cost exceeding 20 billion dollars in the United States.2
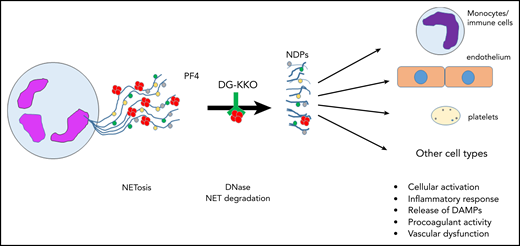
Release of NETs from a neutrophil (left). NETs are decorated with PF4 tetramers (red circles), as well as neutrophil elastase, histones, and myeloperoxidase (colored circles). Degradation of NETs by DNase leads to release of NDPs, which initiate activation of and mediate toxicity toward several cell types. DAMPs, damage-associated molecular patterns; NDPs, NET degradation products.
Release of NETs from a neutrophil (left). NETs are decorated with PF4 tetramers (red circles), as well as neutrophil elastase, histones, and myeloperoxidase (colored circles). Degradation of NETs by DNase leads to release of NDPs, which initiate activation of and mediate toxicity toward several cell types. DAMPs, damage-associated molecular patterns; NDPs, NET degradation products.
First described in 2004,3 NETs have received considerable attention for their role in thromboinflammatory disorders. Initially thought to function simply to “trap” circulating bacteria, the physiologic and pathophysiologic consequences of NET formation and degradation have proven complex and remain incompletely understood (see figure). NETs are extracellular web-like structures consisting of decondensed neutrophil DNA to which proteins derived from neutrophil azurophilic and primary granules, including neutrophil elastase, cathepsin G, myeloperoxidase, and gelatinase, among others, are bound. NETs also bind nuclear proteins, such as histones, that are citrullinated by neutrophil PAD4.4 Release of NETs from neutrophils may occur through specific pathways, such as suicidal, ROS-dependent NETosis, or vital NETosis, in which neutrophils remain viable. Activated platelets may also induce NET formation, a mechanism that may be relevant in sepsis, in which platelets are activated by proteins displaying pathogen-associated molecular patterns recognized by platelet Toll-like receptor 4.5
A critical mechanism underlying the pathogenesis of HIT is FcγRII-mediated platelet activation following binding of heparin-platelet factor 4 (PF4) complexes by HIT antibodies. However, it is now appreciated that HIT is an intensely proinflammatory and prothrombotic disorder associated with complement activation, as well as activation of multiple cell types, including platelets, endothelial cells, and monocytes.6 In a previous study, Gollomp et al also demonstrate that activation of neutrophils leads to the formation of NETs in a passive murine HIT model in which mice are infused with KKO.7 In that study, it was shown that PF4 bound to NETs and that KKO recognized NET-bound PF4 complexes. Binding of PF4, and to an even greater extent, PF4 and KKO, to NETs changed NET morphology by causing NET contraction and impaired the susceptibility of NETs to degradation by DNase. Bound KKO also promoted thrombus formation, presumably by providing an Fc-rich surface to further enhance immune activation of platelets and other cells.
In the current study,1 these investigators used microfluidic approaches and in vivo models to assess the effects of KKO and deglycosylated KKO (DG-KKO) in another thromboinflammatory disorder, sepsis. As in their previous study, PF4 promoted NET contraction and resistance to DNase degradation, with lower levels of NDPs detected in plasma from wild-type mice compared with mice lacking PF4 (cxcl4−/−); the inhibitory effects of PF4 on NET degradation were enhanced in the presence of KKO. NET stabilization by PF4 and KKO also increased bacterial capture. However, despite these seemingly beneficial effects, the presence of the intact Fc region of KKO led to worsened outcomes, including survival, in mice rendered septic in the cecal ligation and puncture (CLP) model; this was attributed to Fc-dependent activation of platelets and immune cells, as well as complement activation. DG-KKO, which has a reduced capacity to activate platelets and fix complement, blocked NET degradation and the release of toxic NET products, but, in contrast to KKO, it improved survival of CLP-treated mice. These studies are important because they provide new information concerning mechanisms of NET-induced toxicity. In particular, they suggest that in sepsis, at least in the murine CLP model, reduction of NDP levels and their well-described systemic toxicities plays an important role in improving outcomes, despite stabilization and persistence of NETs themselves. This hypothesis is consistent with human studies, in which levels of neutrophil-derived circulating cell-free DNA correlate with the multiple organ dysfunction score and other prognostic measurements of sepsis outcomes,8 and levels of circulating DNA-myeloperoxidase complexes correlate with 28-day survival rates in septic patients.9 In addition, Gollomp et al’s data also suggest an additive or synergistic effect of Fc-mediated events, which remain incompletely defined, on the worsening of sepsis outcomes.
Although this compelling study illustrates a novel new approach to therapy of sepsis, there are many questions left unanswered and much additional work that must be performed before clinical application can be further considered. For example, because KKO and DG-KKO cause NET condensation and resistance to degradation, it is essential to better define the mechanisms by which the intact Fc region of KKO leads to worsened outcomes in sepsis models; this might involve activation of platelets, other immune cells, or complement. Moreover, manipulating the formation and degradation of NETs to reduce systemic toxicity in sepsis must balance their ability to trap pathogens against their local toxicities toward endothelial and other cells following neutrophil activation and adhesion, which may be pathogen dependent. Finally, the timing of interventions that modulate NET initiation and degradation seems to be critical, because some studies suggest that administration of DNase to mice soon after induction of CLP may worsen outcomes, whereas later administration reduces inflammation and increases survival.10 Nevertheless, the innovative findings in this study reinforce the proof-of-principle that “hitting back” against the toxic effects of NETs holds promise as a new approach to sepsis.
Conflict-of-interest disclosure: K.R.M. has served as an advisory committee member for Dova, Pfizer, Rigel, and Sanofi. S.S. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal