

Acute erythroid leukemia (AEL) is a unique subtype of acute myeloid leukemia (AML) characterized by erythroid predominance and dysplasia, which is morphologically classified into two subtypes: pure erythroid and myeloid/erythroid leukemias (PEL and MEL). Clinically, AEL exhibits aggressive and rapid clinical course, which is considered to be linked to frequent TP53 mutations and complex karyotypes found in this subtype. Although multiple sequencing studies were conducted to explain such phenotypic variety and to search promising therapeutic targets, neither the diagnostic discrimination of PEL and MEL subtypes nor the establishment of standard therapeutic strategy has fully been successful.
To clarify genetic characteristics and to identify novel therapeutic targets, we comprehensively characterized the mutation and copy number alteration (CNA) profiles and assessed differential impacts of the frequent driver genetic abnormalities on disease phenotypes and clinical outcomes. We initially analyzed 105 cases with AEL (PEL; n=10, MEL; n=69, and unspecified; n=26). Among these, 9 cases were serially analyzed at 2-6 time points. All samples were obtained according to protocols approved by the ethics board of each participating institution. As control, 907 cases with non-erythroid AML (NEL) distinguished by the low fraction (<50%) of erythroid cells in bone marrow were enrolled. Publicly available information on clinical and genetic findings was collected from previous publications, including The Cancer Genome Atlas and BeatAML datasets. In total, 1,012 cases with AML were enrolled in this study. The genetic profile in PEL and MEL were also compared with that of NEL. Finally, we recognized biological significance of the molecular defects by functional analysis of xenograft model, and showed novel therapeutic possibilities targeting pathogenic pathways.
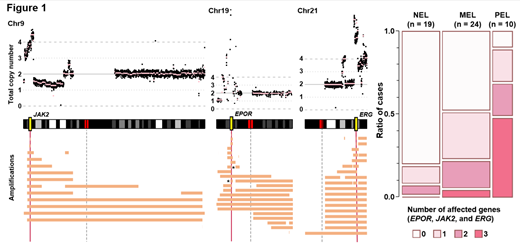
Most frequently observed were mutations in TP53 (39%), STAG2 (20%), and NPM1 (15%), and KMT2A-partial tandem duplication (PTD) (18%) in AEL. On the basis of mutational profiles, consensus clustering divided AEL into 4 subgroups. Group A (n=41, 39%) was defined by TP53 mutations into which all PELs were categorized. In CNA analysis, amplification in 9p, 19p, and 21q were significantly more enriched in PEL than NEL. Intriguingly, amplification of 9p, 19p, and 21q lesions commonly included JAK2, EPOR, and ERG loci, respectively. These genes were affected by amplification or mutations more frequently in AEL than NEL (Figure 1). Group B (n=19, 18%) was enriched for mutations in STAG2, which was significantly more frequent in MEL than in PEL (P<0.05). Compared to STAG2-mutated cases in NEL, those in MEL showed significantly higher frequency of KMT2A-PTD. Group C (n=16, 15%) was defined by NPM1 mutations frequently accompanied by PTPN11 mutations (50% in Group C). This combination was also distinct in MEL and was significantly more frequent than in NPM1-mutated NEL cohort (P<0.05). In Group D (n=29, 28%) without any TP53, STAG2, or NPM1 mutations (triple-negative), RUNX1, ASXL1, BCOR, PHF6, and U2AF1 mutations were enriched compared to triple-negative NEL. In addition, novel loss-of-function USP9X mutations were exclusively identified in Group D AEL (P<0.05, compared to triple-negative NEL), which has been implicated in erythroid leukemogenesis through the TGF beta pathway. Serial sample sequencing analysis revealed that JAK2 mutation positive myeloproliferative neoplasms evolved into PEL. Clinically, patients in Group A showed shorter overall survival time compared to those in any other group. To further search for a novel therapeutic target to the most severe type of AEL, JAK2 inhibitor was administered to xenograft models (from 2 cases) having JAK2 and EPOR amplifications and TP53 mutations. Engrafted cells showed in vitro activation of STAT5 and proliferative capacity enhanced with erythropoietin. In vivo analysis showed that inhibition of JAK/STAT pathway significantly suppressed cell growth and prolonged overall survival.
In summary, our findings suggest that AEL is classified into 4 major subgroups having unique genetic and clinical features. PEL and MEL are genetically distinct subtypes, which was also highlighted by comparison to NEL. Frequent involvement of EPOR/JAK/STAT pathway suggests therapeutic indication of JAK inhibition for AEL especially in the most aggressive type of AEL cases with TP53 mutations.
Yoda:Chordia Therapeutics Inc.: Research Funding. Miyazaki:Kyowa-Kirin: Honoraria; Dainippon-Sumitomo: Honoraria; Nippon-Shinyaku: Honoraria; Chugai: Research Funding; Otsuka: Honoraria; Novartis: Honoraria. Takaori-Kondo:Pfizer: Honoraria; Chugai: Research Funding; Janssen: Honoraria; Kyowa Kirin: Research Funding; Takeda: Research Funding; Ono: Research Funding; Bristol-Myers Squibb: Honoraria, Research Funding; Celgene: Honoraria, Research Funding; Novartis: Honoraria. Nakagawa:Sumitomo Dainippon Pharma Co., Ltd.: Research Funding. Usuki:Astellas Pharma Inc: Research Funding, Speakers Bureau; Daiichi Sankyo Co., Ltd.: Research Funding, Speakers Bureau. Heuser:Bayer Pharma AG, Berlin: Research Funding; Synimmune: Research Funding. Maciejewski:Novartis: Consultancy; Alexion: Consultancy. Ogawa:Qiagen Corporation: Patents & Royalties; RegCell Corporation: Equity Ownership; Kan Research Laboratory, Inc.: Consultancy; ChordiaTherapeutics, Inc.: Consultancy, Equity Ownership; Asahi Genomics: Equity Ownership; Dainippon-Sumitomo Pharmaceutical, Inc.: Research Funding.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal