

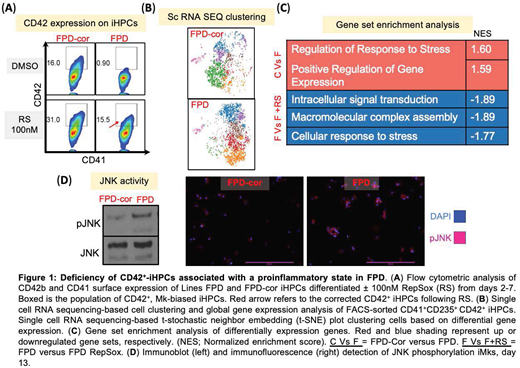
FPDMM is an autosomal dominant disorder associated with qualitative and quantitative platelet defects and an elevated risk of developing acute myeloid leukemia (AML). FPDMM is due to haploinsufficiency of RUNX1, a transcription factor critical for definitive hematopoiesis and megakaryopoiesis. Currently, there are no drugs available for preventing either the bleeding diathesis or AML predisposition in FPDMM. We hypothesize that drugs that correct RUNX1 expression and/or activity in megakaryopoiesis would not only correct the platelet defect, but prevent leukemia progression. In our studies, we established two cell lines with isogenic controls from induced pluripotent stem cells (iPSC) to study the effects of RUNX1 haploinsufficiency in hematopoietic progenitor cells (iHPCs) and megakaryocytes (iMKs). The first iPSC line (FPD) was derived from patient cells harboring a splice mutation in RUNX1 and its isogenic control was created using the CRISPR/Cas9 technology (FPD-cor). The second line was a normal control iPSC line (WT6) in which the FPD splicing mutation was created by CRISPR/Cas9 gene editing (WT6-mut). When differentiated to iMKs, both lines recapitulated a disease phenotype showing decreased iMK yield per iHPC compared to their respective isogenic controls (29% and 42%, respectively). This decreased yield appeared to be due to depletion of a novel CD42+-iHPC subpopulation in both mutant lines compared to their isogenic controls (5% of isogenic controls (Fig. 1A)). Not shown, these HPCs are von Willibrand factor-positive and almost exclusively gives rise to Mks, thus we termed them Mk-biased. To investigate global and local gene expression changes underlying this depletion, single cell RNA sequencing was performed on sorted CD42+-iHPCs derived from the FPD and FPD-cor lines. In both, we defined 7 identical separate subpopulations, several of which were markedly deficient in the FPD CD42+-HPCs (Fig. 1B). Gene set enrichment analysis (GSEA) of genes upregulated in FPD CD42+-iHPCs from the Gene Ontology and Reactome databases indicates that regulation of response to stress and positive regulation of gene expression gene sets are enriched in FPD CD42+-iHPCs (Fig. 1C, shaded in red). Previously, RepSox, a TGFβ receptor 1 inhibitor, was described by others to enhance megakaryopoiesis in humans and mice, and to increase RUNX1 levels. We now show that RepSox can correct the deficient Mk-biased-HPC population (Fig. 1A) as well as return final iMK yield (not shown) to near FPD-cor levels. GSEA of genes downregulated in RepSox-treated FPD CD42+-iHPCs revealed enrichment of stress-response genes(Fig. 1C, shaded in blue). The stress-activated protein kinase c-Jun N-terminal kinase (JNK) is known to be activated by TGFβ1, various cellular stressors, and pro-inflammatory cytokines. Immunoblotting studies of FPD iMKs showed an elevation in baseline JNK2 phosphorylation (p-JNK) levels (Fig. 1D, left), which was not due to alteration in total JNK2 protein compared to isogenic control iMKs. Immunofluorescence microscopy studies supported these findings (Fig. 1D, right). In summary, we describe a novel defect in the inherited platelet disorder FPDMM by studying iPSC-derived hematopoietic cells. We note a marked deficiency of an MK-biased HPC population that may be related to a proinflammatory state in these HPCs potentially involving TGFβ1-related pathways. These findings need to be confirmed in other FPDMM patient iPSCs and show that it also applies to adult hematopoiesis. Whether the described defect is solely related to the observed thrombocytopenia or also is related to the observed AML defect needs also to be addressed.
No relevant conflicts of interest to declare.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal