

Key Points
IKZF1, CDKN2A, ARID5B, and GATA3 influence ALL risk in DS; CDKN2A risk allele penetrance is higher, independent of DS-ALL subtype.
IKZF1 locus impacts enhancer activity and protein binding in a B-cell superenhancer; knockdown leads to more proliferation in DS.

Abstract
Children with Down syndrome (DS) have a 20-fold increased risk of acute lymphoblastic leukemia (ALL) and distinct somatic features, including CRLF2 rearrangement in ∼50% of cases; however, the role of inherited genetic variation in DS-ALL susceptibility is unknown. We report the first genome-wide association study of DS-ALL, comprising a meta-analysis of 4 independent studies, with 542 DS-ALL cases and 1192 DS controls. We identified 4 susceptibility loci at genome-wide significance: rs58923657 near IKZF1 (odds ratio [OR], 2.02; Pmeta = 5.32 × 10−15), rs3731249 in CDKN2A (OR, 3.63; Pmeta = 3.91 × 10−10), rs7090445 in ARID5B (OR, 1.60; Pmeta = 8.44 × 10−9), and rs3781093 in GATA3 (OR, 1.73; Pmeta = 2.89 × 10−8). We performed DS-ALL vs non-DS ALL case-case analyses, comparing risk allele frequencies at these and other established susceptibility loci (BMI1, PIP4K2A, and CEBPE) and found significant association with DS status for CDKN2A (OR, 1.58; Pmeta = 4.1 × 10−4). This association was maintained in separate regression models, both adjusting for and stratifying on CRLF2 overexpression and other molecular subgroups, indicating an increased penetrance of CDKN2A risk alleles in children with DS. Finally, we investigated functional significance of the IKZF1 risk locus, and demonstrated mapping to a B-cell super-enhancer, and risk allele association with decreased enhancer activity and differential protein binding. IKZF1 knockdown resulted in significantly higher proliferation in DS than non-DS lymphoblastoid cell lines. Our findings demonstrate a higher penetrance of the CDKN2A risk locus in DS and serve as a basis for further biological insights into DS-ALL etiology.
Introduction
Down syndrome (DS), which results from partial or complete trisomy of chromosome 21, is one of the most common genetic syndromes and one of the strongest established risk factors for childhood acute leukemia.1,,-4 Compared with children without DS (non-DS), children with DS experience a 10- to 20-fold increased risk of acute leukemia and a 2% lifetime risk of leukemia.5,6 Acute lymphoblastic leukemia (ALL) in children with DS is associated with poorer outcomes and exhibits distinct immunophenotypic and cytogenetic characteristics, including nearly all cases being B lineage (B-ALL) rather than T lineage, higher frequencies of somatic chromosomal rearrangements leading to cytokine receptor-like factor 2 (CRLF2) overexpression and of cooperating Janus kinase 2 (JAK2) activating mutations, a high frequency of somatic IKZF1 deletions,7 and lower frequencies of ETV6-RUNX1 fusion and high hyperdiploidy.6,8,,,,,,,-16 Trisomy 21 clearly predisposes to childhood ALL; however, it is unclear why some children with trisomy 21 develop ALL while others do not.
Genome-wide association studies (GWASs) have identified susceptibility loci for B-ALL in several genes, including IKZF1, CDKN2A, ARID5B, CEBPE, GATA3, BMI1, and PIP4K2A17,,,,,,,,,,,,-30 ; however, germline susceptibility to ALL has not been evaluated specifically in children with DS. Therefore, the primary objectives of this study were to (1) identify inherited genetic variants associated with DS-ALL susceptibility, (2) explore the association between inherited genetic variation and common somatic alterations in childhood ALL, (3) compare the frequency of established ALL risk alleles between ALL cases with and without DS, and (4) investigate the functional implications of DS-ALL susceptibility loci in the genetic background of trisomy 21.
Methods
Study design
The study protocol was approved by the institutional review boards at Baylor College of Medicine, the California Health and Human Services Agency, the University of California (San Francisco and Berkeley), Yale University, and Washington State. This GWAS meta-analysis included 4 independent studies of cases (subjects with DS-ALL) and controls (subjects with DS without a known diagnosis of ALL) (Figure 1): (1) Children’s Oncology Group (COG)/National Down Syndrome Project (NDSP) study 1 (312 DS-ALL cases and 501 DS controls), (2) COG/NDSP study 2 (134 cases and 358 controls), (3) the Michigan-based DS-ALL study (25 cases and 398 controls), and (4) the International Study of DS Acute Leukemia (IS-DSAL) (187 cases [mainly from Childhood Leukemia International Consortium studies]31 and 200 controls). All DS-ALL cases with available immunophenotype data were confirmed B-ALL. Germline DNA was used in all studies for single-nucleotide polymorphism (SNP) genotyping. See supplemental Methods (available on the Blood Web site) for details on recruitment, DNA extraction, and genotyping.
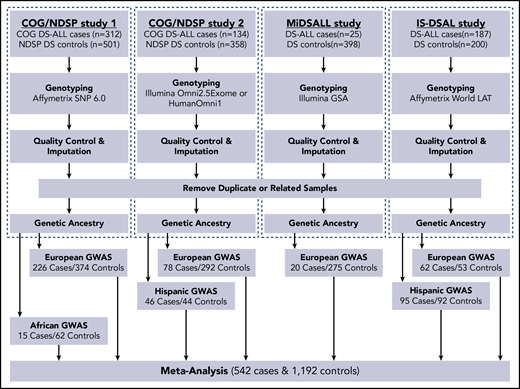
Flowchart of study design and analytic approach. GSA, Global Screening Array; MiDSALL, Michigan-based DS-ALL study.
Flowchart of study design and analytic approach. GSA, Global Screening Array; MiDSALL, Michigan-based DS-ALL study.
Genotype quality control and imputation
Quality control and filtering of genotype data were performed in PLINK v1.9,32 as described in supplemental Methods. Whole-genome imputation of disomic autosomal chromosomes, excluding chromosome 21, was conducted through the Michigan Imputation Server.33 Phasing of genotyped SNPs, which passed quality control, was completed with ShapeIT prior to imputation with Minimac3 using the Haplotype Reference Consortium r1.1 as reference panel.34 Following imputation, genetic variants with a minor allele frequency (MAF) ≥0.01, estimated imputation quality score (R2) >0.30, and Hardy-Weinberg equilibrium P > 1.0 × 10−4 among controls were retained for association analyses.
Chromosome 21 SNPs were called and analyzed separately, as described in supplemental Methods.
Subgroup analysis based on CRLF2 expression
Approximately 50% of DS-ALL cases harbor CRLF2 rearrangements that result in overexpression.15,16 CRLF2 surface expression as determined by flow cytometry (using the cutoff established by the COG flow cytometry central reference labs, namely flow ratio ≥1.2 compared with lymphocytes or >20% thymic stromal lymphopoietin receptor compared with an FMO control) was available for 218 DS-ALL cases (101 Europeans in COG/NDSP study 1 and 75 Europeans and 42 Hispanics in COG/NDSP study 2). It was not possible to perform definitive genetic testing to identify specific CRLF2 rearrangements in the current study, but in another cohort of 279 cases, CRLF2 overexpression by flow as determined by the COG central reference labs demonstrated 100% sensitivity and 100% specificity in identifying cases that had confirmed CRLF2 rearrangements (P2RY8 or IGH) by fluorescence in situ hybridization (Michael Borowitz, Johns Hopkins Univeristy, and Brent Wood, University of Washington, e-mail, 20 June 2019). SNP allele frequencies in DS-ALL cases with CRLF2 overexpression (CRLF2 high) and with normal CRLF2 expression (CRLF2 nl) were separately compared with DS controls from the corresponding study and ancestry groups (n = 769). Results were then meta-analyzed across the 3 groups.
IKZF1 functional assessment
Epigenetic annotation
We used HaploReg v4.1 to determine the regulatory potential of rs58923657 and SNPs in high linkage disequilibrium (LD) (r2 > 0.6 in 1000G EUR reference panel) in lymphoid cells.35 We annotated the haplotype block with respect to cells of B lineage by analyzing chromatin state segmentation, DNase-seq, and transcription factor and histone modification chromatin immunoprecipitation sequencing (ChIP-seq) datasets for the lymphoblastoid cell line (LCL) GM12878 from ENCODE.36 We determined the positions of overlapping and nearby superenhancers in human CD19+ B cells.37 We examined the chromosomal spatial organization at IKZF1 in GM12878 using publicly available datasets from high-throughput chromosome confirmation capture (Hi-C)38 and chromatin interaction analysis by paired-end tag sequencing (ChIA-PET)39 experiments. The positions of chromatin contact domains were called using the Arrowhead algorithm.38
Cell lines and genotyping
Epstein-Barr virus–transformed LCLs were generated using blood samples from DS and non-DS ALL patients treated at Texas Children’s Hospital. Cell lines were maintained in RPMI 1640 with 15% fetal bovine serum (LCLs). Genomic DNA was extracted from low-passage LCLs using Qiagen AllPrep DNA/RNA Mini Kits. Genotypes for IKZF1 SNP rs6964969, the top directly genotyped variant in COG/NDSP study 1 and in perfect LD with rs58923657, were determined using a TaqMan SNP genotyping assay (Assay ID: C__28953799_10; ThermoFisher, Waltham, MA) on a StepOnePlus Real-Time PCR system (ThermoFisher) according to the manufacturer’s instructions.
Luciferase reporter assays
Risk and nonrisk variants of the strong candidate enhancer overlapping rs6964969 and rs58923657 were cloned into the pGL3 Promoter Vector (Promega, Madison, WI). For luciferase assays, DS and non-DS LCLs were electroporated with allele-specific reporter vectors, and enhancer activity was assayed using a Dual-Luciferase Reporter Assay System (Promega). Full details are described in supplemental Methods.
Electrophoretic mobility shift assays
Electrophoretic mobility shift assays (EMSAs) were performed using nuclear extract from DS and non-DS LCLs and allele-specific 5′-IRDye 700–labeled EMSA probes encompassing SNPs rs62445866, rs6964969, rs6944602, rs10264390, and rs17133807 (supplemental Table 5). Binding specificity was assessed by including 10-fold molar excess of unlabeled probe with the same sequence. Samples were resolved by nondenaturing TBE-acrylamide gel electrophoresis (Bio-Rad, Hercules, CA) and imaged with a LI-COR Odyssey Infrared Imaging System (LI-COR Biosciences, Lincoln, NE). Full details are described in supplemental Methods.
Lymphoid proliferation assay
Cells were transduced with lentivirus expressing either IKZF1 or control nontargeting (NT) short hairpin RNA (shRNA) (MISSION pLKO.1-IKZF1-shRNA-puro-GFP and pLKO.1-NT-puro-GFP; MilliporeSigma, St. Louis, MO) by spinfection on retronectin-coated plates (Takara, Mountain View, CA). shRNA vectors were provided by William Carroll.40 Sorted GFP+ LCLs were cultured in triplicate in puromycin-selection medium and counted daily for 5 days. Full methods are described in supplemental Methods.
Statistical analysis
For genome-wide association analyses, we compared genotype dosage of autosomal imputed variants that passed quality control between DS-ALL cases and DS controls using SNPTEST v2.5.4 software, assuming additive allelic effects. Principal components were calculated from directly genotyped SNPs and incorporated as covariates to adjust for genetic ancestry. In each study, genetic ancestry was assessed using STRUCTURE v.2.3.4 software,41 and individuals were assigned to either European, African, Asian, or Hispanic groups, as described in supplemental Methods. Within each study and ancestral group combination, odds ratios (ORs) and corresponding 95% confidence intervals (CIs) were calculated for included variants. We implemented GWAS meta-analysis in METAL software,42 weighting effect estimates by their study- and ancestry-specific standard errors, to obtain overall effect estimates and P values for variants present in at least 6 of the 7 study ancestry-specific populations.
To investigate the risk effects of ALL-associated variants in children with DS relative to non-DS children, we compared allele frequencies of 7 well-replicated ALL GWAS SNPs (in IKZF1, CDKN2A, ARID5B, CEBPE, GATA3, BMI1, and PIP4K2A) in ALL cases (ie, DS-ALL cases vs non-DS ALL cases) and non-ALL controls (DS controls vs non-DS controls). These analyses were performed unadjusted and also adjusted for molecular subgroup, as described in detail in supplemental Methods.
IKZF1 functional data were analyzed using GraphPad Prism. Mean cell concentrations from the lymphoid proliferation assay were regressed as a function of time ± standard error of the mean (SEM) with best-fit lines generated using linear regression. Mean regression slopes for shRNA constructs were compared by 2-tailed t test. Statistical significance was evaluated at α = 0.05.
Results
We meta-analyzed 6 758 624 autosomal and non–chromosome 21 genetic variants across 7 independent DS-ALL case-control study ancestry groups (Figure 1) and report results for SNPs successfully imputed in at least 6 of the populations (Figure 2). Overall, genome-wide significant (P < 5 × 10−8) associations were observed at 4 loci (Table 1), with lead SNPs including rs58923657 near IKZF1 (OR, 2.02; P = 5.32 × 10−15), CDKN2A missense mutation rs3731249 (OR, 3.63; P = 3.91 × 10−10), rs7090445 near ARID5B (OR, 1.60; P = 8.44 × 10−9), and rs3781093 near GATA3 (OR, 1.73; P = 2.89 × 10−8). The direction of effect estimates was largely consistent across study ancestry groups; however, the magnitude of effect for several risk loci varied across groups. For instance, the IKZF1 SNP was more strongly associated with ALL susceptibility among individuals of European (OR, 2.20; 95% CI, 1.80-2.69) and African (OR, 4.06; 95% CI, 1.73-9.52) ancestry than individuals of Hispanic ancestry (OR, 1.28; 95% CI, 0.87-1.90). Similarly, the risk variant in CDKN2A was more strongly associated with ALL among Europeans (OR, 4.22; 95% CI, 2.72-6.54) than Hispanics (OR, 1.56; 95% CI, 0.56-4.39), and was monomorphic in Africans, while GATA3 risk variants exhibited stronger effects among Hispanics (OR, 2.31; 95% CI, 1.64-3.25) and Africans (OR, 3.64; 95% CI, 1.48-8.98) compared with Europeans (OR, 1.50; 95% CI, 1.19-1.89). We also replicated associations at additional previously reported loci (supplemental Table 1), including rs12769953 near BMI1 (OR, 1.37; P = 3.09 × 10−3), rs10741006 near PIP4K2A (OR, 1.48; P = 1.11 × 10−5), rs2239633 near CEBPE (OR, 1.27; P = 5.99 × 10−3), and rs24762284 near ELK3 (OR, 1.20; P = .033).

Manhattan plot of DS-ALL GWAS meta-analysis results. Genome-wide −log10(P) values from meta-analysis of 7 separate DS-ALL GWA studies, including 4 European (in COG/NDSP study 1 and 2, Michigan-based DS-ALL study, and IS-DSAL), 2 Hispanic (in COG/NDSP study 2 and IS-DSAL), and 1 African American ancestry (COG/NDSP study 1) case-control sets. Analysis included 6 758 624 autosomal and non–chromosome 21 SNPs (trisomic genotypes analyzed separately), and results are reported for SNPs successfully imputed in at least 6 out of 7 studies. Red horizontal line represents the genome-wide significance threshold of P = 5 × 10−8.
Manhattan plot of DS-ALL GWAS meta-analysis results. Genome-wide −log10(P) values from meta-analysis of 7 separate DS-ALL GWA studies, including 4 European (in COG/NDSP study 1 and 2, Michigan-based DS-ALL study, and IS-DSAL), 2 Hispanic (in COG/NDSP study 2 and IS-DSAL), and 1 African American ancestry (COG/NDSP study 1) case-control sets. Analysis included 6 758 624 autosomal and non–chromosome 21 SNPs (trisomic genotypes analyzed separately), and results are reported for SNPs successfully imputed in at least 6 out of 7 studies. Red horizontal line represents the genome-wide significance threshold of P = 5 × 10−8.
Genome-wide statistically significant (Pmeta < 5 × 10−8) loci associated with ALL susceptibility in DS by genetic ancestry group and meta-analysis of 4 independent case-control studies
| SNP | Chr | Pos | Gene | European (386 DS-ALL cases, 994 DS controls) | Hispanic (141 DS-ALL cases, 136 DS controls) | African (15 DS-ALL cases, 62 DS controls) | Meta-analysis (542 DS-ALL cases, 1192 DS controls) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | ||||
| rs58923657 | 7 | 50472842 | IKZF1 | 2.20 (1.80-2.69) | 1.18e-14 | 1.28 (0.87-1.90) | .212 | 4.06 (1.73-9.52) | 3.51e-4 | 2.02 (1.70-2.41) | 5.32e-15 |
| rs3731249* | 9 | 21970916 | CDKN2A | 4.22 (2.72-6.54) | 1.20e-10 | 1.56 (0.56-4.39) | .397 | — | — | 3.63 (2.42-5.43) | 3.91e-10 |
| rs7090445 | 10 | 63721176 | ARID5B | 1.50 (1.24-1.80) | 1.92e-5 | 1.90 (1.35-2.67) | 2.43e-4 | 2.45 (1.01-5.99) | .036 | 1.60 (1.36-1.88) | 8.44e-9 |
| rs3781093 | 10 | 8101927 | GATA3 | 1.50 (1.19-1.89) | 5.88e-4 | 2.31 (1.64-3.25) | 1.52e-5 | 3.64 (1.48-8.98) | 9.25e-3 | 1.73 (1.43-2.10) | 2.89e-8 |
| SNP | Chr | Pos | Gene | European (386 DS-ALL cases, 994 DS controls) | Hispanic (141 DS-ALL cases, 136 DS controls) | African (15 DS-ALL cases, 62 DS controls) | Meta-analysis (542 DS-ALL cases, 1192 DS controls) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | ||||
| rs58923657 | 7 | 50472842 | IKZF1 | 2.20 (1.80-2.69) | 1.18e-14 | 1.28 (0.87-1.90) | .212 | 4.06 (1.73-9.52) | 3.51e-4 | 2.02 (1.70-2.41) | 5.32e-15 |
| rs3731249* | 9 | 21970916 | CDKN2A | 4.22 (2.72-6.54) | 1.20e-10 | 1.56 (0.56-4.39) | .397 | — | — | 3.63 (2.42-5.43) | 3.91e-10 |
| rs7090445 | 10 | 63721176 | ARID5B | 1.50 (1.24-1.80) | 1.92e-5 | 1.90 (1.35-2.67) | 2.43e-4 | 2.45 (1.01-5.99) | .036 | 1.60 (1.36-1.88) | 8.44e-9 |
| rs3781093 | 10 | 8101927 | GATA3 | 1.50 (1.19-1.89) | 5.88e-4 | 2.31 (1.64-3.25) | 1.52e-5 | 3.64 (1.48-8.98) | 9.25e-3 | 1.73 (1.43-2.10) | 2.89e-8 |
Chr, chromosome; Pos, base pair position (human genome assembly GRCh37 [hg 19]).
CDKN2A SNP rs3731249 was not assessed in Africans as the minor allele frequency was too low (MAF <0.01) in this population.
Our separate analysis of chromosome 21 SNPs did not reveal any SNPs consistently associated with ALL status across studies following manual inspection of trisomic genotype clusters (results not shown).
Rearrangements of CRLF2 leading to overexpression are the most frequent sentinel genetic alterations in DS-ALL. To identify germline genetic variants specifically associated with CRLF2-overexpressing (CRLF2-high) DS-ALL, we compared 114 CRLF2-high DS-ALL cases to 769 DS controls and 104 CRLF2-normal (nl) cases to the same set of controls. We did not identify any loci associated with CRLF2 status at genome-wide significance (results not shown). The associations between CRLF2 status and 7 previously reported ALL susceptibility loci are summarized in Table 2. The observed effects for most SNPs were largely consistent across CRLF2-high and CRLF2-nl cases with the exception of rs7089424 near ARID5B, which was not associated with CRLF2-high (OR, 1.07, P =.638) but was strongly associated with CRLF2-nl DS-ALL (OR, 2.32; P = 2.95 × 10−7) (test for heterogeneity P value = 5.4 × 10−4). The effect size of the GATA3 SNP rs3824662 was higher in CRLF2-high than CRLF2-nl DS-ALL (OR, 1.72 vs OR, 1.40), as previously reported in non-DS ALL,21 though this difference was not significant (heterogeneity P value = 0.38).
Association between published ALL susceptibility loci and CRLF2 overexpression status in children with DS
| SNP | Chr | Pos | Gene | CRLF2 overexpression (114 DS-ALL cases, 769 DS controls) | No CRLF2 overexpression (104 DS-ALL cases, 769 DS controls) | ||||
|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | Direction* | OR (95% CI) | P | Direction* | ||||
| rs11978267 | 7 | 50466304 | IKZF1 | 2.04 (1.51-2.76) | 3.02e-6 | +++ | 1.93 (1.41-2.64) | 4.37e-5 | +++ |
| rs3731249 | 9 | 21970916 | CDKN2A | 4.27 (2.36-7.71) | 1.49e-6 | +++ | 3.38 (1.71-6.71) | 4.85e-4 | ++− |
| rs3824662 | 10 | 8104208 | GATA3 | 1.72 (1.26-2.35) | 5.97e-4 | +++ | 1.40 (0.99-1.96) | 0.051 | +++ |
| rs12769953 | 10 | 22407656 | BMI1 | 1.45 (0.98-2.14) | 0.061 | +++ | 1.26 (0.85-1.86) | 0.25 | +−+ |
| rs10741006 | 10 | 22856019 | PIP4K2A | 1.27 (0.93-1.71) | 0.13 | +++ | 1.67 (1.17-2.36) | 4.25e-3 | +++ |
| rs7089424 | 10 | 63752159 | ARID5B | 1.07 (0.80-1.45) | 0.64 | −++ | 2.32 (1.68-3.20) | 2.95e-7 | +++ |
| rs2239633 | 14 | 23589057 | CEBPE | 1.46 (1.07-1.98) | 0.017 | +++ | 1.18 (0.85-1.64) | 0.31 | +−− |
| SNP | Chr | Pos | Gene | CRLF2 overexpression (114 DS-ALL cases, 769 DS controls) | No CRLF2 overexpression (104 DS-ALL cases, 769 DS controls) | ||||
|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | Direction* | OR (95% CI) | P | Direction* | ||||
| rs11978267 | 7 | 50466304 | IKZF1 | 2.04 (1.51-2.76) | 3.02e-6 | +++ | 1.93 (1.41-2.64) | 4.37e-5 | +++ |
| rs3731249 | 9 | 21970916 | CDKN2A | 4.27 (2.36-7.71) | 1.49e-6 | +++ | 3.38 (1.71-6.71) | 4.85e-4 | ++− |
| rs3824662 | 10 | 8104208 | GATA3 | 1.72 (1.26-2.35) | 5.97e-4 | +++ | 1.40 (0.99-1.96) | 0.051 | +++ |
| rs12769953 | 10 | 22407656 | BMI1 | 1.45 (0.98-2.14) | 0.061 | +++ | 1.26 (0.85-1.86) | 0.25 | +−+ |
| rs10741006 | 10 | 22856019 | PIP4K2A | 1.27 (0.93-1.71) | 0.13 | +++ | 1.67 (1.17-2.36) | 4.25e-3 | +++ |
| rs7089424 | 10 | 63752159 | ARID5B | 1.07 (0.80-1.45) | 0.64 | −++ | 2.32 (1.68-3.20) | 2.95e-7 | +++ |
| rs2239633 | 14 | 23589057 | CEBPE | 1.46 (1.07-1.98) | 0.017 | +++ | 1.18 (0.85-1.64) | 0.31 | +−− |
Direction of risk allele effects for COG/NDSP study 1 European samples, COG/NDSP study 2 European samples, and COG/NDSP study 2 Hispanic samples.
We next compared the risk allele frequencies of 7 well-replicated ALL susceptibility loci (Table 3) between DS-ALL and non-DS ALL cases. Among 567 DS-ALL cases from our overall study and 3083 non-DS ALL cases in the CCRLP, we found that CDKN2A missense SNP rs3731249 (OR, 1.58; Pmeta = 4.1 × 10−4), GATA3 SNP rs3824662 (OR, 1.34; Pmeta = 4.4 × 10−5), IKZF1 SNP rs11978267 (OR, 1.18; Pmeta = 0.015), and CEBPE SNP rs2239633 (OR, 1.15; Pmeta = 0.043) risk allele frequencies were significantly higher in DS-ALL cases. SNPs in PIP4K2A and BMI1 showed similar trends, whereas ARID5B SNP rs7089424 showed no association with DS status (Pmeta = 0.71). SNP risk allele frequencies did not significantly differ between DS and non-DS controls (supplemental Table 2).
Results from case-case analysis of association between ALL risk alleles and DS status
| SNP | Pos | Gene | DS-ALL CCRLP comparison* (567 DS-ALL, 3083 non-DS ALL) | DS-ALL COG comparison† (255 DS-ALL, 2387 non-DS ALL) | DS-ALL COG molecular subgroup adjusted comparison‡ (255 DS-ALL, 2387 non-DS ALL) | |||
|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | |||
| rs11978267 | Chr7:50466304 | IKZF1 | 1.18 (1.03-1.36) | .015 | 1.15 (0.95-1.39) | .154 | 0.97 (0.77-1.23) | .820 |
| rs3731249 | Chr9:21970916 | CDKN2A | 1.58 (1.23-2.03) | 4.08e-4 | 1.80 (1.27-2.55) | .001 | 1.72 (1.10-2.69) | .017 |
| rs3824662 | Chr10:8104208 | GATA3 | 1.34 (1.16-1.54) | 4.38e-5 | 1.34 (1.09-1.64) | .006 | 0.81 (0.63-1.06) | .121 |
| rs12769953 | Chr10:22407656 | BMI1 | 1.17 (0.98-1.39) | .077 | 1.12 (0.88-1.43) | .343 | 1.12 (0.83-1.50) | .461 |
| rs10741006 | Chr10:22856019 | PIP4K2A | 1.15 (0.99-1.34) | .074 | 1.04 (0.85-1.28) | .691 | 0.96 (0.75-1.23) | .759 |
| rs7089424 | Chr10:63752159 | ARID5B | 0.98 (0.85-1.11) | .71 | 0.77 (0.64-0.93) | .006 | 0.80 (0.64-1.01) | .056 |
| rs2239633 | Chr14:23589057 | CEBPE | 1.15 (1.01-1.32) | .043 | 1.15 (0.94-1.40) | .167 | 1.17 (0.92-1.47) | .199 |
| SNP | Pos | Gene | DS-ALL CCRLP comparison* (567 DS-ALL, 3083 non-DS ALL) | DS-ALL COG comparison† (255 DS-ALL, 2387 non-DS ALL) | DS-ALL COG molecular subgroup adjusted comparison‡ (255 DS-ALL, 2387 non-DS ALL) | |||
|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | |||
| rs11978267 | Chr7:50466304 | IKZF1 | 1.18 (1.03-1.36) | .015 | 1.15 (0.95-1.39) | .154 | 0.97 (0.77-1.23) | .820 |
| rs3731249 | Chr9:21970916 | CDKN2A | 1.58 (1.23-2.03) | 4.08e-4 | 1.80 (1.27-2.55) | .001 | 1.72 (1.10-2.69) | .017 |
| rs3824662 | Chr10:8104208 | GATA3 | 1.34 (1.16-1.54) | 4.38e-5 | 1.34 (1.09-1.64) | .006 | 0.81 (0.63-1.06) | .121 |
| rs12769953 | Chr10:22407656 | BMI1 | 1.17 (0.98-1.39) | .077 | 1.12 (0.88-1.43) | .343 | 1.12 (0.83-1.50) | .461 |
| rs10741006 | Chr10:22856019 | PIP4K2A | 1.15 (0.99-1.34) | .074 | 1.04 (0.85-1.28) | .691 | 0.96 (0.75-1.23) | .759 |
| rs7089424 | Chr10:63752159 | ARID5B | 0.98 (0.85-1.11) | .71 | 0.77 (0.64-0.93) | .006 | 0.80 (0.64-1.01) | .056 |
| rs2239633 | Chr14:23589057 | CEBPE | 1.15 (1.01-1.32) | .043 | 1.15 (0.94-1.40) | .167 | 1.17 (0.92-1.47) | .199 |
Meta-analysis of European (364 DS-ALL, 1154 non-DS ALL) and Hispanic (203 DS-ALL, 1929 non-DS ALL) ALL cases.
Analysis adjusted for top 5 principal components.
Analysis adjusted for top 5 principal components and molecular subgroups (CRLF2 high, high hyperdiploidy, ETV6-RUNX1, and B other).
As the composition of molecular subgroups differs between DS-ALL and non-DS ALL, we next compared SNP risk allele frequencies between a subset of 255 DS-ALL cases and 2387 non-DS B-ALL cases from COG trials P9900 and AALL023227,43 for which subgroup data were available. These COG case-case comparisons were carried out adjusted for (1) genetic ancestry only and (2) for genetic ancestry and molecular subgroups. In case-case analyses adjusted only for genetic ancestry, we found a similar direction and magnitude of effect for each SNP, with the exception of the ARID5B SNP rs7089424, which showed a significant association with non-DS status (OR, 0.77; P = .006) (Table 3). In analyses adjusted for molecular subgroups, the CDKN2A missense SNP rs3731249 remained significantly associated with DS status (OR, 1.72; P = .017), the ARID5B SNP rs7089434 sustained a strong trend toward association with non-DS status (OR, 0.80; P = .056), but differences at other SNPs were largely attenuated.
Finally, we tested for association with DS status within specific ALL molecular subgroups (Table 4). Although not statistically significant within these smaller subgroups, the CDKN2A missense SNP rs3731249 was consistently associated with DS status in all subgroups examined (CRLF2 high, high hyperdiploidy, ETV6-RUNX1, and B other). The ARID5B SNP rs7089424 risk allele was significantly less frequent in DS-ALL than non-DS ALL in the CRLF2-high subgroup (OR, 0.45; 95% CI, 0.30-0.67). As described above, ARID5B SNP rs7089424 was not associated with CRLF2-high ALL risk in DS; however, this variant was strongly associated with CRLF2-high ALL risk in a non-DS case-control analysis (OR, 2.22; 95% CI, 1.65-2.98; supplemental Table 3).
Results from subgroup-specific case-case analysis of association between ALL risk alleles and DS status
| SNP | Position | Gene | CRLF2-high (151 DS-ALL, 55 non-DS ALL) | High hyperdiploid (19 DS-ALL, 888 non-DS ALL) | ETV6-RUNX1 (45 DS-ALL, 547 non-DS ALL) | B-other (40 DS-ALL, 859 non-DS ALL) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | |||
| rs11978267 | Chr7:50466304 | IKZF1 | 0.71 (0.48-1.05) | .08 | 1.26 (0.67-2.36) | .47 | 0.89 (0.55-1.45) | .65 | 1.37 (0.87-2.17) | .17 |
| rs3731249 | Chr9:21970916 | CDKN2A | 2.16 (0.96-4.89) | .06 | 1.62 (0.49-5.43) | .43 | 1.68 (0.64-4.39) | .29 | 1.49 (0.60-3.68) | .39 |
| rs3824662 | Chr10:8104208 | GATA3 | 0.73 (0.50-1.06) | .10 | 1.02 (0.48-2.20) | .95 | 1.02 (0.59-1.78) | .94 | 0.74 (0.42-1.29) | .29 |
| rs12769953 | Chr10:22407656 | BMI1 | 1.28 (0.78-2.11) | .33 | 1.52 (0.58-3.95) | .40 | 0.75 (0.45-1.22) | .24 | 1.45 (0.76-2.77) | .26 |
| rs10741006 | Chr10:22856019 | PIP4K2A | 0.85 (0.56-1.31) | .47 | 0.75 (0.38-1.48) | .41 | 0.78 (0.49-1.23) | .28 | 1.73 (1.00-2.97) | .04 |
| rs7089424 | Chr10:63752159 | ARID5B | 0.45 (0.30-0.67) | 1.0e-4 | 1.38 (0.70-2.74) | .36 | 1.02 (0.65-1.61) | .93 | 1.06 (0.68-1.66) | .80 |
| rs2239633 | Chr14:23589057 | CEBPE | 1.35 (0.93-1.98) | .12 | 0.92 (0.47-1.80) | .81 | 1.19 (0.76-1.88) | .45 | 1.01 (0.62-1.63) | .99 |
| SNP | Position | Gene | CRLF2-high (151 DS-ALL, 55 non-DS ALL) | High hyperdiploid (19 DS-ALL, 888 non-DS ALL) | ETV6-RUNX1 (45 DS-ALL, 547 non-DS ALL) | B-other (40 DS-ALL, 859 non-DS ALL) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | |||
| rs11978267 | Chr7:50466304 | IKZF1 | 0.71 (0.48-1.05) | .08 | 1.26 (0.67-2.36) | .47 | 0.89 (0.55-1.45) | .65 | 1.37 (0.87-2.17) | .17 |
| rs3731249 | Chr9:21970916 | CDKN2A | 2.16 (0.96-4.89) | .06 | 1.62 (0.49-5.43) | .43 | 1.68 (0.64-4.39) | .29 | 1.49 (0.60-3.68) | .39 |
| rs3824662 | Chr10:8104208 | GATA3 | 0.73 (0.50-1.06) | .10 | 1.02 (0.48-2.20) | .95 | 1.02 (0.59-1.78) | .94 | 0.74 (0.42-1.29) | .29 |
| rs12769953 | Chr10:22407656 | BMI1 | 1.28 (0.78-2.11) | .33 | 1.52 (0.58-3.95) | .40 | 0.75 (0.45-1.22) | .24 | 1.45 (0.76-2.77) | .26 |
| rs10741006 | Chr10:22856019 | PIP4K2A | 0.85 (0.56-1.31) | .47 | 0.75 (0.38-1.48) | .41 | 0.78 (0.49-1.23) | .28 | 1.73 (1.00-2.97) | .04 |
| rs7089424 | Chr10:63752159 | ARID5B | 0.45 (0.30-0.67) | 1.0e-4 | 1.38 (0.70-2.74) | .36 | 1.02 (0.65-1.61) | .93 | 1.06 (0.68-1.66) | .80 |
| rs2239633 | Chr14:23589057 | CEBPE | 1.35 (0.93-1.98) | .12 | 0.92 (0.47-1.80) | .81 | 1.19 (0.76-1.88) | .45 | 1.01 (0.62-1.63) | .99 |
Analyses comparing DS-ALL and non-DS ALL cases enrolled on COG P9900 or AALL0232 trials. P values and ORs calculated using logistic regression tests assuming additive allelic effects, adjusting for the top 5 principal components.
Because the IKZF1 risk locus was the top association signal in our DS-ALL GWAS meta-analysis (Figure 2), we explored functional consequences of genetic variation in this locus, which encodes IKAROS, a master regulator of hematopoiesis. Since IKAROS regulates lymphoid differentiation in large part through effects on transcriptional activation and repression networks,44,45 we investigated the epigenetic profile of our top IKZF1 association signal rs58923657. Analysis of ENCODE data for GM12878,36 a non-DS LCL that is homozygous for the rs58923657 nonrisk allele, revealed that rs58923657 maps to an active enhancer and that several SNPs in LD (r2 > 0.6) overlap enhancer histone modifications, DNase I hypersensitive sites, and transcription factor (TF) binding sites (Figure 3A; supplemental Table 4).46,47 Evaluation of publicly available data on chromatin spatial interactions further supported a superenhancer overlapping the IKZF1 risk locus. H3K27ac ChIP-seq data from human CD19+ B cells demonstrated existence of the superenhancer,37 and Hi-C and ChIA-PET data from GM12878 showed that the superenhancer interacts with a distal site upstream of the IKZF1 promoter,38 via a CTCF-looped chromatin structure (Figure 3B).
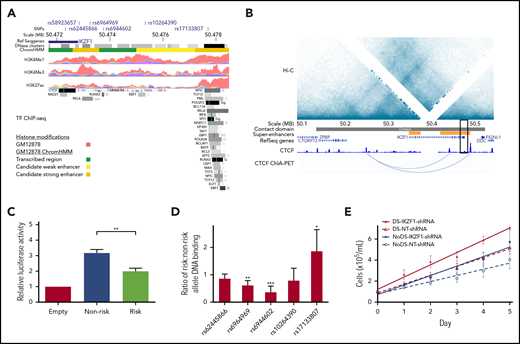
Functional characterization of the IKZF1 susceptibility locus. (A) Epigenetic profile of the IKZF1 SNP rs58923657 susceptibility locus. Tracks show positions of rs58923657 and SNPs in LD (r2 ≥ 0.6); IKZF1; DNase I hypersensitive site clusters; GM12878 chromatin state (ChromHMM) corresponding to transcribed regions (green), candidate weak enhancers (yellow), or candidate strong enhancers (orange); and ChIP-seq data for H3K4Me1, H3K4Me3, H3K27ac and transcription factors from ENCODE.36 Data are displayed in the UCSC genome browser (http://genome.ucsc.edu/)46 and the WashU Epigenome Browser (http://epigenomegateway.wustl.edu/).47 (B) Chromatin spatial organization of chromosome 7 from 50.0-50.6 MB. Tracks show a Hi-C heatmap of chromatin contact frequencies in GM12878; chromatin contact domain determined by the Arrowhead algorithm38 ; CD19+ B-cell superenhancers37 ; canonical transcripts; CTCF ChIP-seq data in GM12878 from ENCODE; and CTCF ChIA-PET mapping data in GM12878.39 Black rectangle indicates the rs58923657 locus. (C) Risk and nonrisk allele-specific enhancers encompassing rs58923657 were cloned into the pGL3-promoter vector (Promega) and transfected into 20 LCLs (10 DS and 10 non-DS). Enhancer reporter assay demonstrates relative luciferase activity measured 24 hours later for the nonrisk and risk alleles relative to the empty vector. Bars show the mean ± SEM from transfections performed in triplicate in the 20 LCLs. (D) EMSAs were performed using nuclear extracts from 6 patient-derived LCLs (3 DS and 3 non-DS) reacted with double-stranded DNA probes encompassing the indicated IKZF1 SNPs. Bars show mean ratios ± SEM of risk to nonrisk allele protein binding. (E) IKZF1 knockdown results in increased cellular proliferation with a greater effect in DS LCLs. Serial cell counts demonstrated significantly greater cellular proliferation for 4 DS LCLs expressing IKZF1-shRNA compared with NT-shRNA (P = .026) and 4 non-DS LCLs expressing IKZF1-shRNA compared with NT-shRNA (P = .017), and the effect of IKZF1-shRNA was significantly greater in the DS LCLs than in the non-DS LCLs (P = .047). Data shown are means of experiments performed in triplicate. P values in panels C-E were determined by a Student 2-tailed t test. *P < .05; **P < .001; ***P < .0001.
Functional characterization of the IKZF1 susceptibility locus. (A) Epigenetic profile of the IKZF1 SNP rs58923657 susceptibility locus. Tracks show positions of rs58923657 and SNPs in LD (r2 ≥ 0.6); IKZF1; DNase I hypersensitive site clusters; GM12878 chromatin state (ChromHMM) corresponding to transcribed regions (green), candidate weak enhancers (yellow), or candidate strong enhancers (orange); and ChIP-seq data for H3K4Me1, H3K4Me3, H3K27ac and transcription factors from ENCODE.36 Data are displayed in the UCSC genome browser (http://genome.ucsc.edu/)46 and the WashU Epigenome Browser (http://epigenomegateway.wustl.edu/).47 (B) Chromatin spatial organization of chromosome 7 from 50.0-50.6 MB. Tracks show a Hi-C heatmap of chromatin contact frequencies in GM12878; chromatin contact domain determined by the Arrowhead algorithm38 ; CD19+ B-cell superenhancers37 ; canonical transcripts; CTCF ChIP-seq data in GM12878 from ENCODE; and CTCF ChIA-PET mapping data in GM12878.39 Black rectangle indicates the rs58923657 locus. (C) Risk and nonrisk allele-specific enhancers encompassing rs58923657 were cloned into the pGL3-promoter vector (Promega) and transfected into 20 LCLs (10 DS and 10 non-DS). Enhancer reporter assay demonstrates relative luciferase activity measured 24 hours later for the nonrisk and risk alleles relative to the empty vector. Bars show the mean ± SEM from transfections performed in triplicate in the 20 LCLs. (D) EMSAs were performed using nuclear extracts from 6 patient-derived LCLs (3 DS and 3 non-DS) reacted with double-stranded DNA probes encompassing the indicated IKZF1 SNPs. Bars show mean ratios ± SEM of risk to nonrisk allele protein binding. (E) IKZF1 knockdown results in increased cellular proliferation with a greater effect in DS LCLs. Serial cell counts demonstrated significantly greater cellular proliferation for 4 DS LCLs expressing IKZF1-shRNA compared with NT-shRNA (P = .026) and 4 non-DS LCLs expressing IKZF1-shRNA compared with NT-shRNA (P = .017), and the effect of IKZF1-shRNA was significantly greater in the DS LCLs than in the non-DS LCLs (P = .047). Data shown are means of experiments performed in triplicate. P values in panels C-E were determined by a Student 2-tailed t test. *P < .05; **P < .001; ***P < .0001.
Next, we examined allele-specific effects of rs58923657 on enhancer activity using a luciferase reporter in patient-derived DS and non-DS LCLs and found that the risk allele was associated with significantly decreased enhancer activity compared with the nonrisk allele in all LCLs (P < .001, Figure 3C; supplemental Figure 1). We also investigated differences in protein-DNA interactions at 5 candidate functional SNPs, at which proteins are reported to bind in LCLs (supplemental Table 4), in the rs58923657 haplotype block using EMSAs (supplemental Table 5). Concordant with the decreased enhancer activity of the rs58923657 risk allele, we also observed significantly decreased protein binding to the risk allele in nuclear extracts from 6 LCLs (3 DS and 3 non-DS) for rs6964969 (P < .001) and rs6944602 (P < .0001) (Figure 3D; supplemental Figure 2), which overlap TF binding sites for RELA and RUNX3, respectively (Figure 3A). Conversely, we observed significantly increased nuclear protein binding to the risk allele for rs17133807 (P < .05) (Figure 3D; supplemental Figure 2), which overlaps a DNase I hypersensitive site that binds many TFs (Figure 3A). We did not observe any significant differences between DS and non-DS LCLs in either enhancer activity or DNA-protein binding (data not shown).
Last, since ALL-associated SNPs in IKZF1 have been previously associated with lower IKZF1 expression levels,20 we investigated the effects of IKZF1-shRNA knockdown on proliferation in DS and non-DS LCLs homozygous for the nonrisk haplotype. IKZF1 knockdown resulted in an average of 66% decrease in IKAROS protein expression across the transduced LCLs (supplemental Figure 3) and led to significantly higher rates of cellular proliferation as measured by serial cell counts in both DS (P = .026) and non-DS LCLs (P =.017) compared with NT-shRNA controls (Figure 3E). Of note, the magnitude of effect of IKZF1 knockdown on proliferation was greater in the DS genetic background, with a significantly higher rate of proliferation in DS-IKZF1-shRNA LCLs compared with non-DS-IKZF1-shRNA LCLs (P = .047).
Discussion
We performed the first GWAS of ALL risk in children with DS to investigate genetic loci that might contribute toward the 10- to 20-fold increased risk and distinctive somatic cytogenetic spectrum of ALL in this population.5,6 This study of 542 DS-ALL cases and 1192 DS controls demonstrates that established non-DS ALL susceptibility loci also contribute to ALL risk in children with DS. Specifically, we identified genome-wide significant association signals at loci near IKZF1, CDKN2A, ARID5B, and GATA3, and replicated associations (P < .05) at loci near BMI1, PIP4K2A, CEBPE, and a recently identified locus at ELK3. Considered collectively, our findings suggest that rather than unique loci influencing ALL risk in this highly susceptible population, trisomy 21 appears to modify the penetrance of inherited ALL susceptibility, particularly for the CDKN2A risk locus.
Although we did not identify novel ALL susceptibility loci, the pattern of SNP associations observed in DS-ALL was distinctive in several respects. First, the magnitude of the effect size was greater for several loci than reported previously in non-DS ALL, including IKZF1, GATA3, and CDKN2A. Of particular note, the CDKN2A missense mutation rs3731249 conferred a ∼3.6-fold increased risk of DS-ALL (and a >4-fold risk in Europeans) compared with reported effect sizes of ∼2.2 to 3 in non-DS children.29,30,48 The apparent increased penetrance of these ALL risk SNPs in DS children was further supported by their significant associations with DS status in case-case comparisons. To some extent, these results appear to reflect differences in cytogenetic subgroup frequencies between DS-ALL and non-DS ALL,9,10,14 as case-case associations were largely attenuated after adjusting for subgroup. However, the CDKN2A risk locus remained significantly associated with DS status in the adjusted case-case analysis (OR, 1.72), and its effect was consistent across subgroups, suggesting that this locus exerts a stronger effect in the background of trisomy 21. Within the specific molecular subgroup subanalyses, the ARID5B SNP rs7089424 demonstrated the most significant difference in allele frequency between DS-ALL and non-DS ALL cases in the CRLF2-high subgroup. This discrepancy was largely due to the lack of association between ARID5B and CRLF2-high DS-ALL, contrary to what was observed in non-DS ALL. In CRLF2-nl DS-ALL cases, the effect estimate for the ARID5B risk allele (OR, 2.32) was similar to that reported for non-DS ALL.20,23,49 Additional work is warranted to elucidate the potential modifying effects of trisomy 21 on the association between the ARID5B risk allele and CRLF2 rearrangements.
As previously described,27 we identified some differences in risk allele effect estimates across ancestral groups. For example, the association with variation in GATA3 was stronger in Hispanics, where risk allele frequency was previously shown to correlate with increased levels of Native American ancestry among Hispanic ALL patients,50 leading to a higher risk allele frequency in this population (eg, rs3824662 [GATA3] MAF 0.23 in HapMap Europeans vs MAF 0.42 in HapMap Mexicans). The stronger effect of the GATA3 locus in Hispanics may also reflect an increased frequency of the Ph-like ALL subtype in this population.21,51 Conversely, the risk loci in IKZF1 and CDKN2A are more frequent among individuals of European ancestry and were more strongly associated with DS-ALL in Europeans than Hispanics. Regardless of differences in individual SNP effect estimates across ancestral groups, we found a similar enrichment of CDKN2A missense SNP rs3731249 risk alleles in DS-ALL compared with non-DS ALL.
The top association signal identified in our DS-ALL GWAS was in a locus near IKZF1, and in silico analyses revealed this region maps to a putative B-cell superenhancer that is involved in long-range chromatin interactions with a region proximal to the IKZF1 promoter, suggesting that germline variation in this locus could impact lymphoid enhancer activity associated with IKZF1 expression. Functional assessment of the IKZF1 risk locus showed that the IKZF1 risk haplotype is associated with significantly decreased enhancer activity and affects protein binding at several SNPs in the haplotype block. Our finding that the IKZF1 ALL risk haplotype exhibits reduced enhancer activity constitutes a novel insight into the mechanism underlying this susceptibility locus, and is consistent with the association of the risk allele with reduced IKAROS expression and with the known importance of IKAROS for normal lymphoid lineage transcriptional priming and differentiation.20,44 Further functional studies are necessary, including cellular assays where the trisomy 21 environment may play a role, to fully characterize the effects associated with variation in this haplotype block.
Since variants in this region are associated with decreased IKZF1 expression,20 we also investigated functional effects of decreased IKZF1 expression in isogenic backgrounds, by shRNA knockdown in DS and non-DS LCLs homozygous for the nonrisk allele. Knockdown of IKZF1 led to increased proliferation, supporting a proleukemogenic effect of the IKZF1 risk allele. Moreover, this effect was stronger in the DS genetic background, providing mechanistic support for the increased effect of the IKZF1 locus on ALL susceptibility in children with vs those without DS. Given that we did not see differences in the effects of IKZF1 risk alleles on enhancer activity in DS vs non-DS LCLs, this suggests that a similar reduction in IKZF1 expression may lead to increased ALL risk in children with DS, perhaps in conjunction with the effects of trisomy 21 on hematopoiesis. Additional work is needed to determine whether IKZF1 somatic alterations are less frequent in DS-ALL cases harboring germline IKZF1 risk alleles or if there is preferential somatic loss of the protective allele in cases with 1 copy of the risk allele, as demonstrated previously for the CDKN2A missense SNP.29,30
Our findings should be interpreted in light of some limitations. This study may have been underpowered to identify novel susceptibility loci with modest effects. This is particularly true for potential associations in non-European populations and associations with specific genetic subgroups, since data on cytogenetics and CRLF2 expression were only available for a subset of DS-ALL cases. The use of a heterogeneous sample with respect to somatic profiles may dilute statistical power to identify subtype-specific variants; however, the >500 cases and 1100 controls included in this study represent the largest assessment of ALL risk in a DS population to date. Additionally, we were limited in our ability to systematically evaluate chromosome 21 variants in children with DS using SNP array data. A novel association with ALL was recently reported at the chromosome 21 gene ERG in Hispanics.52 Our chromosome 21 SNP analysis could not determine an association at ERG with DS-ALL; however, a parallel study of Hispanic DS-ALL cases and controls did identify an association using targeted SNP genotyping.53 Alternative approaches, such as next-generation sequencing of chromosome 21 in DS-ALL cases and controls, may provide additional insights into the role of trisomy 21 on ALL risk.
This study provides evidence of a higher penetrance of the CDKN2A missense variant rs3731249 in children with trisomy 21. Additionally, our findings support trisomy 21 disruption of the expression of transcriptional activators involved in lymphoid hematopoiesis, leading to the dysregulation of B-lymphoid progenitor cell development.54,55 Given that children with DS have a 10- to 20-fold increased risk of developing ALL, this study provides valuable insight into the impact of common inherited genetic variation on ALL risk in children with DS. The particularly strong association between DS-ALL and the relatively infrequent (global MAF <0.01) CDKN2A missense variant rs3731249 (ORmeta = 3.63; 95% CI, 2.42-5.43) may have implications for future surveillance and genetic counseling in children with DS. Indeed, the use of common polymorphisms for genetic risk prediction in ALL has been hampered by moderate effect sizes, high-risk allele frequencies, and the low background rate of ALL in the general population, all of which combine to limit positive predictive value. Given its large effect size and low risk allele frequency, CDKN2A missense SNP rs3731249 may overcome these limitations when applied to populations with high rates of ALL, such as children with DS, though such analyses may require multicenter prospective analyses of children with DS. Future, large-scale, collaborative studies of well-phenotyped cases are needed to further elucidate the role of inherited genetic variation in leukemia susceptibility in children with DS, including next-generation sequencing studies to detect additional low-frequency variants with large effect sizes, which may significantly contribute to ALL risk on an individual level.
For original data, please contact Karen R. Rabin at krrabin@texaschildrens.or.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Amos Gaikwad and Tatiana Goltsova for their assistance with flow cytometry analyses. The authors also thank Robin Cooley and Steve Graham (Genetic Disease Screening Program, California Department of Public Health) for their assistance and expertise in the procurement and management of DBS specimens. The authors are also grateful to the Washington State Department of Health for additional specimen/data access and to William O’Brien of the University of Washington for programming/data management. The authors additionally thank the families for their participation in the California Childhood Leukemia Study (formerly known as the Northern California Childhood Leukemia Study). For recruitment of subjects enrolled in the California Childhood Leukemia Study, the authors gratefully acknowledge the clinical investigators at the following collaborating hospitals: University of California, Davis Medical Center (Jonathan Ducore); University of California, San Francisco (Mignon Loh and Katherine Matthay); Children’s Hospital of Central California (Vonda Crouse); Lucile Packard Children’s Hospital (Gary Dahl); Children’s Hospital Oakland (James Feusner and Carla Golden); Kaiser Permanente Roseville (formerly Sacramento) (Kent Jolly and Vincent Kiley); Kaiser Permanente Santa Clara (Carolyn Russo, Alan Wong, and Denah Taggart); Kaiser Permanente San Francisco (Kenneth Leung); Kaiser Permanente Oakland (Daniel Kronish and Stacy Month); California Pacific Medical Center (Louise Lo); Cedars-Sinai Medical Center (Fataneh Majlessipour); Children’s Hospital Los Angeles (Cecilia Fu); Children’s Hospital Orange County (Leonard Sender); Kaiser Permanente Los Angeles (Robert Cooper); Miller Children’s Hospital Long Beach (Amanda Termuhlen); University of California, San Diego Rady Children’s Hospital (William Roberts); and University of California, Los Angeles Mattel Children’s Hospital (Theodore Moore).
This work was supported by grant RP170074 from the Cancer Prevention and Research Institute of Texas (K.R.R. and P.J.L.) and by funds from the Jeffrey Pride Foundation and the COG Foundation (K.R.R.). Work was also supported in part by the National Institutes of Health National Cancer Institute (NCI) (grant K07 CA218362 to A.L.B.). Flow cytometry assays were performed at the Research Flow Cytometry Core Facility of Texas Children’s Cancer and Hematology Centers with the support from the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the NCI (cancer center support grant P30CA125123). The work of St. Jude investigators is supported by NCI grants CA21765 and P50 GM115279 and the American Lebanese Syrian Associated Charities. The work of COG investigators was supported by the National Institutes of Health NCI (grants U10 CA 29139, U10 CA98543, U10 CA98413, U10 CA180886, and U10 CA180899 to the COG) and supported by St. Baldrick’s Foundation. S.P.H. is the Jeffrey E. Perelman Distinguished Chair in the Department of Pediatrics at The Children's Hospital of Philadelphia. The IS-DSAL study was supported by Alex’s Lemonade Stand Foundation “A” Awards (A.J.d.S. and K.M.W.), the Emerging Investigator Fellowship Grant from the Pediatric Cancer Research Foundation (A.J.d.S.), The Children’s Health and Discovery Initiative of Translating Duke Health (K.M.W.), and the National Institutes of Health NCI (research grants R01 CA155461 to J.L.W. and X.M.) and National Institute of Environmental Health Sciences (R01 ES009137 to C.M., P24 ES004705 to C.M., and R24 ES028524 to C.M. and L.M.). The 2016-2019 Childhood Leukemia International Consortium Scientific Annual Meetings were supported in part by the National Institutes of Health, National Institute of Environmental Health Sciences (award number U13 ES026496).
The IS-DSAL study included biospecimens and/or data obtained from the California Biobank Program (screening information system requests 26 and 572), Section 6555(b), 17 California code of regulations. The California Department of Public Health is not responsible for the results or conclusions drawn by the authors of this publication. The collection of cancer incidence data used in the CCRLP study was supported by the California Department of Public Health pursuant to California Health and Safety Code Section 103885, Centers for Disease Control and Prevention’s (CDC) National Program of Cancer Registries, under cooperative agreement 5NU58DP003862-04/DP003862, the National Cancer Institute’s Surveillance, Epidemiology and End Results Program under contract HHSN261201000140C awarded to the Cancer Prevention Institute of California, contract HHSN261201000035C awarded to the University of Southern California, and contract HHSN261201000034C awarded to the Public Health Institute. The ideas and opinions expressed herein are those of the author(s) and do not necessarily reflect the opinions of the State of California, Department of Public Health, the National Institutes of Health, and the Centers for Disease Control and Prevention or their Contractors and Subcontractors. This study used birth data obtained from the State of California Center for Health Statistics and Informatics. The California Department of Public Health is not responsible for the analyses, interpretations, or conclusions drawn by the authors regarding the birth data used in this publication.
Authorship
Contribution: K.R.R., P.J.L., A.J.d.S., and J.J.Y. designed the study; A.L.B., A.J.d.S., V.U.G., W.Y., J.J.Y., P.J.L., and K.R.R. prepared the manuscript; V.U.G., A.L.B., A.J.d.S., N.A.K., S.A.P., G.E.D., E.A.E, L.F.B., and H.M.H. performed experiments; A.L.B., A.J.d.S., V.U.G., W.Y., K.M.W., J.M.C., E.F., I.S., M.E.S., A.T.D., J.J.Y., P.J.L., K.R.R., N.W., N.A.H., A.J.C. M.J.B., B.L.W., E.F., M.E.Z., M.D., and M.V.R. analyzed data; W.L.C., E.A.R., L.M., A.Y.K., J.H., C.L., D.S., J.W.T., J.M.B., P.T., L.G.S., M.S.P., S.P.H., C.-H.P., C.G.M., M.L.L., C.M., X.M., B.A.M., S.L.S., J.L.W., P.J.L., and K.R.R. provided patient samples; and all authors edited and approved the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Philip J. Lupo, Department of Pediatrics, Baylor College of Medicine, One Baylor Plaza, MS: BCM305, Houston, TX 77030; e-mail: philip.lupo@bcm.edu; and Karen R. Rabin, Department of Pediatrics, Baylor College of Medicine, 1102 Bates St, Suite 750.00, Houston, TX 77030; e-mail: krrabin@texaschildrens.org.
