

Key Points
CRISPR-induced truncation of the FBXW7 substrate binding domain identifies cleaved NOTCH1 (NICD) as a target of FBXW7 in CLL cells.
FBXW7 mutations in CLL correlate with increased NICD protein and elevated NOTCH1 target gene expression and thereby mimic NOTCH1 mutations.

Abstract
NOTCH1 is mutated in 10% of chronic lymphocytic leukemia (CLL) patients and is associated with poor outcome. However, NOTCH1 activation is identified in approximately one-half of CLL cases even in the absence of NOTCH1 mutations. Hence, there appear to be additional factors responsible for the impairment of NOTCH1 degradation. E3-ubiquitin ligase F-box and WD40 repeat domain containing-7 (FBXW7), a negative regulator of NOTCH1, is mutated in 2% to 6% of CLL patients. The functional consequences of these mutations in CLL are unknown. We found heterozygous FBXW7 mutations in 36 of 905 (4%) untreated CLL patients. The majority were missense mutations (78%) that mostly affected the WD40 substrate binding domain; 10% of mutations occurred in the first exon of the α-isoform. To identify target proteins of FBXW7 in CLL, we truncated the WD40 domain in CLL cell line HG-3 via clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein-9 (Cas9). Homozygous truncation of FBXW7 resulted in an increase of activated NOTCH1 intracellular domain (NICD) and c-MYC protein levels as well as elevated hypoxia-inducible factor 1-α activity. In silico modeling predicted that novel mutations G423V and W425C in the FBXW7-WD40 domain change the binding of protein substrates. This differential binding was confirmed via coimmunoprecipitation of overexpressed FBXW7 and NOTCH1. In primary CLL cells harboring FBXW7 mutations, activated NICD levels were increased and remained stable upon translation inhibition. FBXW7 mutations coincided with an increase in NOTCH1 target gene expression and explain a proportion of patients characterized by dysregulated NOTCH1 signaling.
Introduction
Mutational analysis of cancer DNA identifies candidate oncogenes and tumor-suppressor genes. However, the functional role of these candidate genes frequently remains unknown. In chronic lymphocytic leukemia (CLL), leukemogenic genes are recurrently affected by genetic mutations1 that can, in the case of TP53 and NOTCH1, even predict response to therapy.2
Novel mutations have been identified at lower frequencies3 but can still point to central cellular pathways affected in the cancer cells. One of the genes affected by such mutations is F-box and WD40 repeat domain containing-7 (FBXW7). FBXW7 encodes an E3-ubiquitin ligase with 3 isoforms α, β, and γ that differ only in their N-termini due to alternative first exon usage and in subcellular localization. All 3 isoforms share the dimerization domain, the F-Box that binds to the ubiquitin-degradation complex and the WD40 substrate binding domain. The WD40 domain interacts with proteins that are subsequently subject to proteasomal degradation.4,5
FBXW7 targets proto-oncoproteins such as cleaved NOTCH1 (NOTCH1 intracellular domain [NICD]), c-MYC, NF-κB2/p100, Cyclin E, and hypoxia-inducible factor 1-α (HIF1-α) for proteasomal degradation.6-13 These substrates are recognized by the FBXW7-WD40 binding domain that includes the conserved amino acid residues R465, R479, and R505,14 which are essential for high-affinity substrate binding.15,16 These 3 residues are hotspots prone to harbor mutations in several cancer entities.4 Mutations in FBXW7 have been identified in hematopoietic tumors like T-cell acute lymphoblastic leukemia (T-ALL; 30%) as well as in solid tumors. In T-ALL, NOTCH1/FBXW7 mutations correlate with a better prognosis17 in the absence of mutations/deletions of PTEN. In contrast, in solid tumors FBXW7 mutations correlate with cancer progression and resistance to treatment.18,19 In CLL patients, FBXW7 mutations occur at a frequency of 2% to 6% and commonly affect the WD40 substrate binding domain.1,20-23 FBXW7 mutations are associated with trisomy 12 in the mature CD19+ B-cell CLL cells20 and both the activating NOTCH1 and FBXW7 mutations are thought to contribute to the transformation of CLL to an aggressive lymphoma also known as Richter syndrome.21 However, the molecular consequences of FBXW7 mutations in CLL have so far not been investigated.
Mutations in FBXW7 could influence its impact on target proteins that are also of relevance in CLL. CLL cells receive prosurvival signals from the microenvironment in the blood, lymph nodes, and bone marrow. The microenvironment can activate NF-κB signaling, induce HIF1-α24 and stimulate cyclin-dependent proliferation.25 NOTCH1 is mutated in 10% of untreated CLL cases and in up to 20% of progressive/relapsed CLL patients. Mutations of NOTCH1 result in activation of targets like c-MYC.3,26,27
To understand the functional consequences of FBXW7 mutations and their contribution to the dysregulation of NOTCH1 signaling, we generated clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein-9 (Cas9) engineered CLL cell lines with heterozygous/homozygous truncation of the FBXW7-WD40 domain and identified an accumulation of NICD protein and increased NOTCH1 activity. These findings were confirmed in CLL patient cells harboring FBXW7 mutations. We found an increase in NICD levels that were stable even after the inhibition of translation. Additionally, we confirmed a reduction in NICD binding to FBXW7 harboring CLL-specific mutations by coimmunoprecipitation. Furthermore, expression of NOTCH1 target genes was elevated in FBXW7-mutated CLL cases similar to that in patients with NOTCH1 mutations. We postulate that CLL with FBXW7 mutations can be functionally included into the group of CLL patients with dysregulated NOTCH1 signaling.
Methods
Detailed materials and protocols for amplicon-based targeted next-generation sequencing, cell-culture conditions, reverse transcription–quantitative polymerase chain reaction (RT-qPCR) analysis, western blotting, coimmunoprecipitation, plasmids, transfection, and luciferase reporter assays are available as supplemental Methods (available on the Blood Web site).
Generation of HG-3 cell lines with FBXW7 mutations via CRISPR/Cas9 technology
The CRISPR/Cas9 vector pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (MIT, Cambridge, MA) (Addgene plasmid #48138).28 Four different guide RNAs specific to FBXW7 were screened and the oligo TGTGGTAACCGAATAGTTAG TGG was chosen for further transfection experiments in HG-3 CLL cells. Single positive cells were selected via flow cytometry and expanded as clonal homogeneous cell populations. Genomic DNA was isolated from expanded clones (Qiagen) and indel mutations within the WD40 domain were confirmed by Sanger sequencing.
Statistics
Statistical analysis was performed with GraphPad Prism. One-way analysis of variance followed by Bonferroni’s posttest or Kruskal-Wallis followed by Dunn’s multiple comparison test was used to determine statistical significance (****P < .0001; ***P < .001; **P < .01; *P < .05).
Results
In CLL, FBXW7 missense mutations most frequently affect the WD40 substrate binding domain
To unravel which functional domain and isoform of FBXW7 is most commonly targeted by mutations in CLL patients, we sequenced CD19-sorted CLL cells from a cohort of 905 previously untreated patients. Forty-one FBXW7 mutations were identified in 36 of 905 patients (4%) (Figure 1A; supplemental Table 1). All mutations were heterozygous, which has previously been associated with a dominant-negative effect.18 Missense mutations were most commonly found (32 of 41 [78%]), followed by nonsense mutations (4 of 41 [10%]), indel at the nuclear localization signal (NLS) of the FBXW7α-isoform (4 of 41 [10%]), and a splice site mutation (1 of 41 [2%]). Seventy-three percent of FBXW7 mutations were localized within the WD40 substrate binding domain, similar to other cancer entities.4 Thirty-six of the identified 41 mutations (88%) affected all 3 FBXW7 isoforms, whereas 5 mutations (12%) were specific to the α-isoform. Interestingly, 5 of 36 patients (14%) harbored 2 different types of FBXW7 mutations (V514F/R505C, R465C/T15VR, R465C/G423V, R465C/R689W, Q492X/D480N), suggesting a specific selective pressure, and 5 of 36 cases (14%) with FBXW7 mutations had additional NOTCH1 mutations. Amino acid substitutions in FBXW7 at the hotspot arginine residues R465, R479, and R505 affect substrate recognition29 and were identified in 49% of all FBXW7-mutated CLL cases. Hence, we further investigated whether FBXW7 mutations could lead to reduced oncoprotein target recognition in CLL patients.

Identification of FBXW7 mutations in untreated CLL patients. (A) In a total of 905 untreated CLL patients, 41 FBXW7 mutations were identified. The 3 different isoforms of FBXW7 have different N-termini (red boxes) but share the same dimerization (DD; green box), F-Box (blue box), and WD40 domains (purple box). Amino acid residues are numbered for the FBXW7α-isoform (AA (α)). Mutations are denoted by dots colored according to the alteration of the respective amino acid. (B) FBXW7α-isoform specific indel mutation result in the T15VR mutation within the nuclear localization signal (NLS). (C) Expression of the FBXW7α, -β, and -γ isoforms in 72 CLL patient samples relative to 3 housekeeping genes HPRT1, PPIA, and TBP. mRNA, messenger RNA.
Identification of FBXW7 mutations in untreated CLL patients. (A) In a total of 905 untreated CLL patients, 41 FBXW7 mutations were identified. The 3 different isoforms of FBXW7 have different N-termini (red boxes) but share the same dimerization (DD; green box), F-Box (blue box), and WD40 domains (purple box). Amino acid residues are numbered for the FBXW7α-isoform (AA (α)). Mutations are denoted by dots colored according to the alteration of the respective amino acid. (B) FBXW7α-isoform specific indel mutation result in the T15VR mutation within the nuclear localization signal (NLS). (C) Expression of the FBXW7α, -β, and -γ isoforms in 72 CLL patient samples relative to 3 housekeeping genes HPRT1, PPIA, and TBP. mRNA, messenger RNA.
Although the majority of mutations are shared by all 3 isoforms of FBXW7, 12% affect the FBXW7α-isoform only, of which 80% lead to the insertion/deletion (indel) resulting in amino acid p.(Gly16delinsValArg; here referred to as T15VR) (Figure 1A-B). To understand which FBXW7 isoform is most relevant for the pathogenesis of CLL, we quantified expression of the α-, β-, and γ-isoform in a cohort of CLL patients with untreated and advanced disease. FBXW7A was 1.7-fold more strongly expressed than FBXW7G and even 1000-fold more than FBXW7B (Figure 1C).
In conclusion, FBXW7 mutations are identified in 4% of untreated CLL patients. Mutations are exclusively heterozygotic and frequently affect hotspots in the WD40 domain, suggesting a dominant-negative change of function of FBXW7 in CLL cells.
Common FBXW7 mutations in CLL cause a change in hydrophobicity and electrostatic interactions
To delineate the potential changes in substrate recognition, we analyzed the most common FBXW7 mutations in the WD40 substrate interaction domain (56%) of our CLL cohort (Figure 1A): the hotspot mutations R465C, R479Q, R505C, and G423V as well as W425C and A503V which are in close proximity to these mutational hotspots. We postulate that these mutations affect their interaction with substrates that could act as proto-oncoproteins in CLL. The FBXW7-WD40 domain (residues 263-707) consists of multiple cross β-sheets in a circular conformation and is connected to α-helices extending below (Figure 2A shown in green and blue). The most commonly mutated amino acid residues within the WD40 domain are highlighted, G423 (black), R465 (gray), R479 (blue), and R505 (red) and the less common mutations W425C (purple) and A503V (yellow) in Figure 2B. Intriguingly, these mutated residues are pointing to the exterior of the WD40 β-sheet domain (Figure 2C) and are hence in close proximity to substrate proteins interacting with FBXW7. Mutations leading to amino acid changes G423V, W425C, R465C, R479Q, and R505C are predicted to result in a change of hydrophobic (Figure 2D) as well as electrostatic (Figure 2E) surface interactions of FBXW7 and are therefore likely to change the substrate binding. In addition, the surface contact area with substrates is also reduced due to shortening of sidechains. Note that A503 makes no direct contacts with the substrate but is nearby to the substrate interacting with R479 and Y519 (supplemental Figure 5A-B). However, the A503V mutation may disrupt the local structure slightly and affect substrate binding.

Most common FBXW7 mutations in CLL lead to structural changes in hotspot substrate binding areas resulting in different hydrophobicity and electrostatic interactions. (A) FBXW7 ribbon structure depicting α-helical and β-sheet structures. WD40 domain highlighted in green. (B) Magnification of panel (A) of amino acid residues 423 (black), 425 (purple), 465 (gray), 479 (blue), 503 (yellow), and 505 (red). (C) Partial view from the midpoint of the structure (see black line and arrowheads in panel B) reveals side-chain protrusions of these amino acid residues into the center of the circular β-sheet configuration. Most common mutations cause changes of (D) hydrophobicity and (E) electrostatic surfaces. In panel D, red indicates hydrophobic surfaces; blue, hydrophilic surfaces. In panel E, red indicates negative electrostatic surfaces; blue, positive electrostatic surfaces.
Most common FBXW7 mutations in CLL lead to structural changes in hotspot substrate binding areas resulting in different hydrophobicity and electrostatic interactions. (A) FBXW7 ribbon structure depicting α-helical and β-sheet structures. WD40 domain highlighted in green. (B) Magnification of panel (A) of amino acid residues 423 (black), 425 (purple), 465 (gray), 479 (blue), 503 (yellow), and 505 (red). (C) Partial view from the midpoint of the structure (see black line and arrowheads in panel B) reveals side-chain protrusions of these amino acid residues into the center of the circular β-sheet configuration. Most common mutations cause changes of (D) hydrophobicity and (E) electrostatic surfaces. In panel D, red indicates hydrophobic surfaces; blue, hydrophilic surfaces. In panel E, red indicates negative electrostatic surfaces; blue, positive electrostatic surfaces.
We postulate that based on the low number of truncating FBXW7 mutations (10%) in comparison with missense mutations (78%), which most frequently affect the WD40 substrate binding domain (73%), FBXW7 mutations probably do not abrogate but rather change substrate recognition due to changes in electrostatic and hydrophobic interactions at the WD40 substrate binding domain.
CLL cell lines do not harbor mutations in FBXW7
The in-depth characterization of the molecular function of FBXW7 in CLL requires a lymphoblastoid cell line harboring FBXW7 mutations. To identify such a cell line, we screened CLL (n = 8), mantle cell lymphoma (MCL; n = 5), T-ALL (n = 2), Burkitt lymphoma (n = 1), and lymphoblastoid cell lines (n = 3) for mutations in FBXW7 and additional recurrently mutated genes in CLL (supplemental Table 2). FBXW7 mutations could only be detected in the 2 T-ALL cell lines as previously described.19 In summary, none of the 8 analyzed CLL cell lines exhibited a mutation in FBXW7. Therefore, we modified FBXW7 in the HG-3 CLL cell line to determine the impact on its substrates.
CRISPR/Cas9-induced FBXW7 truncation in the HG-3 cell line reveals NOTCH1, c-MYC, and HIF1-α as relevant substrates in CLL
We introduced a frameshift mutation in the HG-3 CLL cell line that led to the truncation of the WD40 domain of all 3 FBXW7 isoforms (Figure 3A; supplemental Table 3). Of note, the HG-3 cell line does not contain a NOTCH1 mutation (supplemental Table 2). Two clones with homozygous deletion of the FBXW7-WD40 domain (HG-3 D8 and HG-3 D40), as well as 2 clones with heterozygous deletion (HG-3 D13 and HG-3 D24), were selected for further analysis (supplemental Table 3).

Lack of the FBXW7-WD40 domain results in the accumulation of FBXW7 substrates NICD and c-MYC. (A) Heterozygous or homozygous indel mutations were induced in FBXW7 at the protein amino acid position 396 in the CLL cell line HG-3 using CRISPR-Cas9. (B) FBXW7, Cyclin E, p100/p52, c-MYC, and NICD protein levels were analyzed in FBXW7-mutated HG-3 cell lines via western blot (representative of 3 blots). (C) c-MYC promoter activity (n = 3) and (D) transcription factor activity of NOTCH1 (n = 3) was quantified in HG-3 wt and HG-3 clones with FBXW7 truncation using luciferase reporter plasmids with the respective transcription factor binding motifs. Overexpression of the NOTCH1 coactivator RBP was used as positive control. Statistical significance was assessed via 1-way analysis of variance followed by Bonferroni’s posttest (****P < .0001; ***P < .001; *P < .05).
Lack of the FBXW7-WD40 domain results in the accumulation of FBXW7 substrates NICD and c-MYC. (A) Heterozygous or homozygous indel mutations were induced in FBXW7 at the protein amino acid position 396 in the CLL cell line HG-3 using CRISPR-Cas9. (B) FBXW7, Cyclin E, p100/p52, c-MYC, and NICD protein levels were analyzed in FBXW7-mutated HG-3 cell lines via western blot (representative of 3 blots). (C) c-MYC promoter activity (n = 3) and (D) transcription factor activity of NOTCH1 (n = 3) was quantified in HG-3 wt and HG-3 clones with FBXW7 truncation using luciferase reporter plasmids with the respective transcription factor binding motifs. Overexpression of the NOTCH1 coactivator RBP was used as positive control. Statistical significance was assessed via 1-way analysis of variance followed by Bonferroni’s posttest (****P < .0001; ***P < .001; *P < .05).
FBXW7 protein was detectable in the wild-type HG-3 (HG-3 WT) and in the heterozygously mutated cell lines, whereas FBXW7 protein was not detectable in the homozygously mutated cell lines (Figure 3B). This is to be expected because the antibody used for western blotting specifically detects the FBXW7-WD40 domain. Subsequently, proteins that are putative substrates of FBXW7 in CLL cells were analyzed. Cyclin E30,31 and NF-κB2/p10032,33 are well-described substrates of FBXW7. However, no changes in the levels of Cyclin E nor NF-κB2/p100 or its processed protein product p52 were found upon truncation of the WD40 domain. In contrast, increased protein levels of the FBXW7 substrates cleaved NOTCH1 (NICD) and c-MYC were detected upon truncation of FBXW7 (HG-3 D8 and D40; Figure 3B). This is notable as NOTCH1 and c-MYC are of pathogenic relevance in CLL.3,24,26,27 In the heterozygous FBXW7-truncated HG-3 cell lines D13 and D24, only a slight or no difference in NICD and c-MYC protein levels was detected in comparison with the wild-type cell line (Figure 3B).
To investigate whether increased protein levels of FBXW7 substrates would have functional implications, we performed luciferase reporter assays to quantify the transcriptional activity of HIF1-α (supplemental Figure 1) and NOTCH1 as well as the activity of the MYC gene promoter (Figure 3D). In fact, the homozygous truncation of FBXW7 in CLL cells led to a significantly increased HIF1-α activity under hypoxic, but not normoxic, conditions (supplemental Figure 1). An increase in c-MYC gene promoter activity and NOTCH1 transcriptional activity under normoxic conditions was also detectable in these cell lines upon modulation of FBXW7 (Figure 3C-D, cell lines HG-3 D8 and HG-3 D40 in comparison with the FBXW7 WT). Of note, activity of HIF1-α, c-MYC, and NOTCH1 was similar between the heterozygously truncated FBXW7-WD40 cell lines (D13 and D24) and the FBXW7 WT cell line. These findings indicate that heterozygous truncating mutations do not lead to a gain or change of function. The increased protein levels and activity of MYC gene promoter and NOTCH1 as well as the increased HIF1-α activity in FBXW7-WD40 truncated cell lines strongly suggests that these proteins are bona fide substrates of FBXW7 in CLL cells.
NICD levels are increased in primary CLL cells with FBXW7 mutation
Next, we analyzed the abundance of c-MYC and NICD in primary CLL cells that harbor a FBXW7 mutation. Similar to previous results, HIF1-α could not be detected in primary CLL cells from peripheral blood under normoxic conditions.24 However, NICD levels were elevated in primary CLL cells from 4 patients harboring FBXW7 missense mutations (G423V, R465C, T15VR, R465C), supporting our hypothesis that FBXW7 change-of-function mutations lead to protein accumulation of specific substrates (Figure 4A). In line with previous reports,34 cells from 1 of 2 available patients with NOTCH1 mutations but without FBXW7 mutation had elevated NICD levels (Figure 4A). In our study, 2 of 4 patients with FBXW7 mutation displayed elevated NICD levels despite having no additional NOTCH1 gene mutations, suggesting an impact of FBXW7 mutations on NICD stability. Interestingly, high c-MYC levels were not specific to FBXW7 or NOTCH1 mutation (Figure 4B), but might also be influenced by other factors. To assess the turnover of FBXW7 target proteins, we determined the stability of NICD and c-MYC in cells from 4 patients with FBXW7 and/or NOTCH1 mutations in comparison with patient cells without FBXW7 or NOTCH1 mutations (Figure 4C). Whereas c-MYC levels diminished at similar rates in FBXW7-mutated or unmutated patient cells, NICD levels in cells from 5 patients with FBXW7 and/or NOTCH1 mutation were stable even 11 hours after cycloheximide treatment. These findings identified NICD to be consistently dysregulated in FBXW7-mutated CLL patient cells.
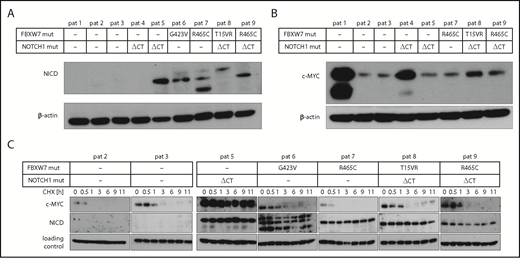
CLL patients with FBXW7 mutations have increased NICD levels. (A) Cleaved NOTCH1 (NICD) and (B) c-MYC protein levels were determined in primary CLL cells from patients with and without FBXW7 mutations. Different NICD band sizes of patients 7 and 8 might be due to degradation products of NICD or due to different posttranslational modifications. (C) Translation in primary CLL patient cells was inhibited by 10 µg/mL cycloheximide (CHX) for the indicated periods of time. NICD and c-MYC protein levels were analyzed via western blotting. Stable c-MYC levels in patient 5 might be observable due to MYC lesions or dysregulation of other proteins responsible for c-MYC degradation. Loading control for patients 2, 5, 6, and 9 is β-actin, for patients 3, 7, and 8 the loading control is α-tubulin. NOTCH1 ΔCT mut, NOTCH1 deletion of a CT dinucleotide (P2514fs); pat, patient.
CLL patients with FBXW7 mutations have increased NICD levels. (A) Cleaved NOTCH1 (NICD) and (B) c-MYC protein levels were determined in primary CLL cells from patients with and without FBXW7 mutations. Different NICD band sizes of patients 7 and 8 might be due to degradation products of NICD or due to different posttranslational modifications. (C) Translation in primary CLL patient cells was inhibited by 10 µg/mL cycloheximide (CHX) for the indicated periods of time. NICD and c-MYC protein levels were analyzed via western blotting. Stable c-MYC levels in patient 5 might be observable due to MYC lesions or dysregulation of other proteins responsible for c-MYC degradation. Loading control for patients 2, 5, 6, and 9 is β-actin, for patients 3, 7, and 8 the loading control is α-tubulin. NOTCH1 ΔCT mut, NOTCH1 deletion of a CT dinucleotide (P2514fs); pat, patient.
Mutations in FBXW7-WD40 can be categorized into NICD binding and nonbinding
Because we identified NICD to be the protein most consistently dysregulated in CLL with mutated FBXW7, we tested whether the CLL-specific FBXW7 mutations would have an impact on NICD binding. To this end, we selected FBXW7 variants harboring mutations within the WD40 domain (G423V, R465C, R479Q, Y545C, W425C, D480N, A503V, V514F, R505C), the α-specific mutations T15VR and V154I as well as the previously detected mutation G16ins18 and compared them to a truncating mutation in close proximity to the WD40 domain (1-362; D362X). Whereas α-specific mutations within the NLS did not affect translocation to the nucleus and still enabled NICD binding (Figure 5; supplemental Figures 2 and 3), truncation of the FBXW7-WD40 domain ablated NICD binding (Figure 5A, lane 7), which is in line with increased NICD levels in the homozygously FBXW7-truncated HG-3 cell lines (Figure 3B). Please note that the truncating mutation D362X shows less expression and/or stability. These results indicate that mutations that alter the WD40-binding domain are probably affecting FBXW7 substrate binding, but do not exclude that mutations outside of this domain might coincide with elevated FBXW7 degradation or messenger RNA silencing.

Effect of patient-specific mutations in FBXW7 on NICD binding. (A-C) Coimmunoprecipitation (CoIP) of NOTCH1-IC (NICD) with wild-type or mutant FBXW7 constructs. HEK293 cells were cotransfected with NICD (which corresponds to the human NOTCH1 intracellular domain, aa 1761-2555) and the indicated Flag-tagged FBXW7α constructs. Expression of NICD (middle panel) and Flag-FBXW7 proteins (bottom panel) was verified by western blotting. CoIPs were performed 24 hours after transfection. *The heavy chain of anti-Flag antibody used for IP. Localization of the corresponding mutations is shown schematically in the topmost panel. (B) Lane 8: expression control for FBXW7, lysate from FBXW7 transfected cells, no-CoIP. Control CoIPs for testing unspecific binding to the Flag column are shown in supplemental Figure 2. hs-NICD, homo sapiens-NICD; IP, immunoprecipitation; WB, western blot.
Effect of patient-specific mutations in FBXW7 on NICD binding. (A-C) Coimmunoprecipitation (CoIP) of NOTCH1-IC (NICD) with wild-type or mutant FBXW7 constructs. HEK293 cells were cotransfected with NICD (which corresponds to the human NOTCH1 intracellular domain, aa 1761-2555) and the indicated Flag-tagged FBXW7α constructs. Expression of NICD (middle panel) and Flag-FBXW7 proteins (bottom panel) was verified by western blotting. CoIPs were performed 24 hours after transfection. *The heavy chain of anti-Flag antibody used for IP. Localization of the corresponding mutations is shown schematically in the topmost panel. (B) Lane 8: expression control for FBXW7, lysate from FBXW7 transfected cells, no-CoIP. Control CoIPs for testing unspecific binding to the Flag column are shown in supplemental Figure 2. hs-NICD, homo sapiens-NICD; IP, immunoprecipitation; WB, western blot.
Interestingly, in silico modeling predicted reduced binding of FBXW7 substrate by a novel hotspot mutation in CLL G423V (found in 3 of 905 CLL cases) as well as by W425C, similar to the hotspot mutations R465C, R479Q, and R505C (Figure 2). We confirmed the lack of binding of NICD to FBXW7 harboring any of these mutations by coimmunoprecipitation (Figure 5; supplemental Figure 2). NICD binding was not affected by the A503V mutation, whereas the other mutations D480N, V514F, Y545C decreased but did not ablate NICD binding (Figure 5; supplemental Figure 2). This retention of binding of NICD by FBXW7 was predicted by the in silico modeling (supplemental Figure 5C-E). Interestingly, the mutations D480N and V514F were identified to co-occur with an additional FBXW7 mutation in CLL (D480N and Q492X; V514F and R505C). In addition, all FBXW7 mutants still localize to the nucleus (supplemental Figure 3) and are able to dimerize with wild-type FBXW7 (supplemental Figure 4), further strengthening the hypothesis of a dominant-negative change-of-function mechanism.
Hence, we were able to identify the FBXW7 mutations G423V and W425C as novel CLL-specific mutations that ablate NICD binding similarly to the common hotspot mutations that affect the residues R465, R479, and R505. Mutations D480N, V514F, and Y545C, which were identified in a total of 3 patients, did not or not completely abolish NICD binding and neither did the α-specific mutations of FBXW7. Thus, in summary, the majority of FBXW7 missense mutations detected in the WD40 domain resulted in decreased NICD binding and therefore suggest that these FBXW7 mutations impair the NOTCH1-signaling pathway in CLL.
NOTCH1 target genes are increased in primary CLL cells with FBXW7 mutation
We then tested expression of NOTCH1 target genes in FBXW7-mutated CLL patient cells. Therefore, we selected 35 bona fide NOTCH1 target genes that have been identified by chromatin immunoprecipitation sequencing in primary CLL cells.35,36 To validate these 35 NOTCH1 target genes, we quantified their expression in CLL tumors and compared patient samples harboring a NOTCH1 mutation (n = 28) to patients with a FBXW7 mutation (n = 5) and patients without NOTCH1 or FBXW7 mutations (n = 174) (Figure 6A; supplemental Table 4). Twenty-three of the postulated NOTCH1 target genes showed no significant differential expression in our data set. However, 11 genes showed an increase in expression in CLL cells with a NOTCH1 mutation compared with cells without a NOTCH1 mutation (ZMIZ1, PTGER4, IL6R, ATF5, NT5E, CD27, CHI3L2, ENTPD1, and UBL7) and PLAC8 was the only gene significantly different in FBXW7-mutated samples, probably due to the low number of affected patients (n = 5; supplemental Table 4). Of these genes, NT5E, CD27, CHI3L2, and PTGER4 displayed the highest fold change in expression. We validated the differential expression of these genes and that of PLAC8 via RT-qPCR in additional patients with a mutation of NOTCH1 (n = 12) or FBXW7 (n = 12) in comparison to patients without mutation in FBXW7 and NOTCH1 (n = 14) and patients with both FBXW7 and NOTCH1 mutations (n = 3). PLAC8 and CD27 were significantly increased and expression of NT5E and CHI3L2 was elevated in NOTCH1- and FBXW7-mutated cases whereas PTGER4 was not differentially expressed (Figure 6B; supplemental Figure 6). Intriguingly, when separating FBXW7 mutations into hotspot mutations (R465, R479, R505, G423V) and other mutations, the fold change of PLAC8 and NT5E expression was even higher in the FBXW7 hotspot mutated samples (Figure 6B). Furthermore, 3 CLL patients harboring both FBXW7 and NOTCH1 mutations expressed similar levels of PLAC8, NT5E, or CHI3L2 compared with that of NOTCH1 and FBXW7 hotspot mutated samples (Figure 6B; supplemental Figure 6).

FBXW7 mutations result in increased NOTCH1 target gene expression.PLAC8, NT5E, and CD27 expression was analyzed in (A) a cohort of NOTCH1 mutated (n = 28), FBXW7 mutated (n = 5), and non-NOTCH1/non-FBXW7 mutated (n = 174) CLL patients via gene expression analysis and (B) validated in another cohort of NOTCH1 mutated (n = 12), FBXW7 mutated (n = 12), NOTCH1 and FBXW7 mutated (n = 3), or nonmutated (n = 14) CLL cases via RT-qPCR. Details about NOTCH1 and FBXW7 mutations are listed in supplemental Table 5. Statistical significance was assessed via the Kruskal-Wallis test followed by the Dunn’s multiple comparison test (**P < .01; *P < .05). GEP, gene expression profiling.
FBXW7 mutations result in increased NOTCH1 target gene expression.PLAC8, NT5E, and CD27 expression was analyzed in (A) a cohort of NOTCH1 mutated (n = 28), FBXW7 mutated (n = 5), and non-NOTCH1/non-FBXW7 mutated (n = 174) CLL patients via gene expression analysis and (B) validated in another cohort of NOTCH1 mutated (n = 12), FBXW7 mutated (n = 12), NOTCH1 and FBXW7 mutated (n = 3), or nonmutated (n = 14) CLL cases via RT-qPCR. Details about NOTCH1 and FBXW7 mutations are listed in supplemental Table 5. Statistical significance was assessed via the Kruskal-Wallis test followed by the Dunn’s multiple comparison test (**P < .01; *P < .05). GEP, gene expression profiling.
Taken together, FBXW7 mutations in CLL lead to an increase not only in NICD levels but also in NOTCH1 target gene expression. Additionally, we have identified novel FBXW7 mutations that occur in CLL. Of these mutations, G423V and W425C ablate NICD binding and hence lead to NICD stabilization, whereas NICD binding is not impaired in cases with α-specific mutations of FBXW7 (T15VR, V154I) and in the presence of the mutation A503V in close proximity to the hotspot residue R505 (Figure 7).
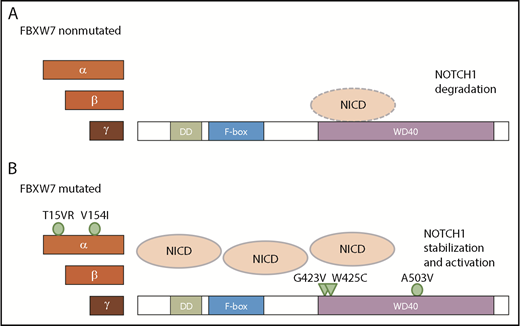
Proposed model of NICD degradation in CLL patients with and without FBXW7 mutations. NICD is targeted by wt FBXW7 (A), whereas mutations in the FBXW7 substrate binding domain G423V and W425C result in insufficient recognition of NICD (B; triangles). In contrast, mutations A503V as well as α-specific mutations T15VR and V154I do not impact on NICD binding (B; circle).
Proposed model of NICD degradation in CLL patients with and without FBXW7 mutations. NICD is targeted by wt FBXW7 (A), whereas mutations in the FBXW7 substrate binding domain G423V and W425C result in insufficient recognition of NICD (B; triangles). In contrast, mutations A503V as well as α-specific mutations T15VR and V154I do not impact on NICD binding (B; circle).
Discussion
Mutations in FBXW7 are recurrent in CLL and are dominant change-of-function mutations
Gene mutations uncover a central role of tumor suppressors and oncogenes. However, their functional relevance frequently remains unclear. In CLL, mutations in FBXW7 have been identified with a high frequency of missense and rare nonsense mutations both affecting the WD40 substrate binding domain (Figure 1A). Here, we are the first to identify the functional consequence of FBXW7 mutations in CLL.
As no CLL cell line could be identified with a FBXW7 mutation (supplemental Table 2), we introduced a truncation of the WD40 substrate binding domain of FBXW7 in the CLL cell line HG-3. We chose to truncate FBXW7 instead of introducing CLL-specific mutations due to very low efficiency of CRISPR/Cas9 in CLL cells. The HG-3 cell lines with a heterozygously truncated FBXW7 did not show clear effects of a dominant change-of-function phenotype. Unfortunately, we could not detect the truncated FBXW7 protein in the HG-3 lines because the antibody used recognizes the FBXW7 C-terminus. However, the truncated FBXW7 could have a decreased stability, which was observed upon recombinant overexpression of a truncated FBXW7 (Figure 5A, see lower panel, lane 7). This also provides an explanation of why, in primary CLL, FBXW7 missense mutations occur more frequently than truncating mutations. Although HG-3 does not fully reflect typical CLL due to its inherent characteristics as a cell line, the homozygous truncation of FBXW7 allowed us to identify substrates specific to FBXW7, which we were able to validate in primary CLL patient cells with heterozygous FBXW7 missense mutations, suggesting they have a dominant-negative effect. Two of the FBXW7 substrates Cyclin E, involved in cell-cycle progression, and NF-κB2/p100 from the noncanonical NF-κB pathway, were not affected by the loss of FBXW7 function in the FBXW7-mutated CLL cell lines (Figure 3B). Although Cyclin E and NF-κB2/p100 have been shown to be of relevance in CLL, their regulation in CLL is likely not affected via FBXW7. Indeed, Cyclin E expression is regulated by miR15/miR16 and NF-κB signaling by BAFF/APRIL receptors through the CLL microenvironment.37,38 In the present work, homozygous truncation of the FBXW7-WD40 domain resulted in the accumulation and increased transcription factor activity of the substrates HIF1-α and NOTCH1 as well as elevated c-MYC promoter activity, pointing to these proteins as targets of FBXW7 in CLL.
All FBXW7 mutations identified in primary CLL patients were heterozygous (Figure 1A). If CLL cells would be at an advantage in fitness due to a loss of function of FBXW7, we would have expected a sizeable proportion of homozygous deletions or truncating mutations of the gene in CLL cells, which was not the case. The proportion of heterozygous FBXW7 mutations rather suggests that the identified missense mutations are most likely change-of-function mutations.
How do mutations define substrate specificity of FBXW7?
Increased NICD protein levels were detectable not only in 1 of 2 CLL patients with a NOTCH1 mutation, but also in 4 patients with missense mutations in the FBXW7-WD40 domain (Figure 4A). NICD levels in primary CLL cells with FBXW7 mutations were even stable 11 hours after inhibition of translation (Figure 4C). Furthermore, we detected an increase in NOTCH1 target gene expression in FBXW7-mutated cases (n = 12) similar to NOTCH1-mutated (n = 12) or NOTCH1/FBXW7-mutated patients (n = 3) in comparison with nonmutated (n = 14) CLL samples (Figure 6B). These findings show that FBXW7 mutations induce NOTCH1 signaling. In contrast, levels of c-MYC, which is a transcriptional target of NOTCH1, were similar in FBXW7-mutated and unmutated primary CLL cells, suggesting additional layers of regulation of c-MYC39 or different modes of binding (Figure 4B; supplemental Figure 7). It should be noted that despite the analysis of NOTCH1 gene expression in our cohort, based on the gene sets of Fabbri et al35 and Ryan et al,36 NICD activity was not assessed in our samples. Yet, we were able to detect significant changes in NOTCH1 target gene expression in our FBXW7- and NOTCH1-mutated samples.
We postulate that NICD dimers cannot bind sufficiently to the WD40 substrate binding domain of mutated FBXW7, hindering ubiquitin transfer (supplemental Figure 7). This is based on our findings that (a) NICD protein levels were increased in FBXW7-mutated patients (Figure 4A), (b) NICD is stable in FBXW7-mutated patient cells even after inhibition of translation (Figure 4C), and (c) NOTCH1 target gene expression is enhanced in FBXW7-mutated CLL cases similarly to NOTCH1-mutated CLL cases (Figure 6). These findings all point to a role of FBXW7 mutations in activating the leukemogenic NOTCH1 pathway. In CLL, both FBXW7 and NOTCH1 mutations correlate with trisomy 12 and the mutational load of either gene correlates with overall survival, a resemblance that further underlines their functional similarity.40
It should be noted that the change of hydrophobic and electrostatic interactions in the FBXW7-WD40 substrate binding domain (Figure 2) may also result in the recognition and binding of yet unknown substrates in CLL cells, leading to their ubiquitination and degradation that could lead to disease progression and possibly also to treatment resistance.
NOTCH1 mutations have been implicated to be of prognostic relevance in CLL.27,41-43 Yet, CLL cells from approximately one-half of patients display elevated NICD protein even in the absence of NOTCH1 mutations.35,41 In this study, we show for the first time that in CLL cells with mutated FBXW7, NICD levels are upregulated and stable, leading to increased expression of NOTCH1 target genes. We suggest that recurrent missense mutations as well as the previously nondescribed FBXW7 mutations G423V and W425C impair substrate binding and targeting, which we can also show for the hotspot mutations in residues R465, R479, and R505. In addition, we were able to show that the mutation A503V that is in close proximity to the hotspot mutation R505C, as well as the α-specific FBXW7 mutations T15VR and V154I, do not impair binding to NICD (Figures 5 and 7). However, mutations A503V, T15VR, and V154I might have an impact on other, yet unknown, FBXW7 substrates that could be of relevance for CLL pathogenesis. In addition, we cannot exclude that the sensitivity of our coimmunoprecipitation experiments might not be sufficient to detect slightly different changes in NICD binding.
Taken together, our data show that FBXW7 mutations reduce NICD binding and degradation and lead to its activation in CLL cells. We expect that the patient subgroup with FBXW7-WD40 mutations will share similar clinical phenotypes to that of patients harboring a NOTCH1 mutation, a hypothesis that will be tested in the future and, due to the predictive nature of NOTCH1 activation in CLL, can help clinical management of the disease.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Karin Müller, Sabrina Schrell, Christina Galler, and Sabrina Rau from the Department of Internal Medicine III, and Sabine Schirmer and Roswitha Rittelmann from the Department of Internal Medicine I, Ulm University for their technical assistance. The authors thank Rhett Kovall for helpful discussions. In addition, the authors are very grateful to the CLL patients for their generous donation of primary tumor material. V.C. and W.C. thank the International Graduate School in Molecular Medicine Ulm at Ulm University, Germany for its support.
This work was supported by the German José Carreras Leukemia Foundation (DJCLS) 14R/2016, German Research Foundation (DFG)-SFB1074/B2, ERA-NET TRANSCAN2 “Fighting Resistance in CLL” (FIRE-CLL), Federal Ministry of Education and Research in Germany (BMBF) e:Bio “Pretherapeutic Epigenetic CLL Patient Stratification” (PRECiSe) (D.M. and S.S.) and DFG-GRK2254, DFG-SFB1074/A3 (F.O.).
Authorship
Contribution: V.C. conducted the experiments; W.C. performed the structural modeling; E.T. performed the amplicon based next-generation sequencing of the patients and cell lines, and analyzed the sequencing results; D.Y.Y. selected and processed peripheral blood mononuclear cell samples of CLL patients; L.P. performed interaction assays; J.B. performed gene-expression profiling and analyzed data; M.-A.W. supplied essential materials for this study; V.C., W.C., S.J.K., M.R., D.Y.Y., L.P., J.B., E.T., M.-A.W., H.D., D.M., S.S., and F.O. discussed and analyzed the data; and V.C., W.C., F.O., and D.M. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Daniel Mertens, Department of Internal Medicine III, University Hospital Ulm, Albert-Einstein-Allee 23, 89081 Ulm, Germany; e-mail: daniel.mertens@uniklinik-ulm.de.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal