

Key Points
FLNA mutations lead to the absence of FLNa in patient MKs and a defect in proplatelet formation via RhoA overactivation.
The increased RhoA activity in FLNA-mutated MKs is driven by the disruption of FLNa/αIIbβ3 interaction.

Abstract
Filamin A (FLNa) links the cell membrane with the cytoskeleton and is central in several cellular processes. Heterozygous mutations in the X-linked FLNA gene are associated with a large spectrum of conditions, including macrothrombocytopenia, called filaminopathies. Using an isogenic pluripotent stem cell model derived from patients, we show that the absence of the FLNa protein in megakaryocytes (MKs) leads to their incomplete maturation, particularly the inability to produce proplatelets. Reduction in proplatelet formation potential is associated with a defect in actomyosin contractility, which results from inappropriate RhoA activation. This dysregulated RhoA activation was observed when MKs were plated on fibrinogen but not on other matrices (fibronectin, vitronectin, collagen 1, and von Willebrand factor), strongly suggesting a role for FLNa/αIIbβ3 interaction in the downregulation of RhoA activity. This was confirmed by experiments based on the overexpression of FLNa mutants deleted in the αIIbβ3-binding domain and the RhoA-interacting domain, respectively. Finally, pharmacological inhibition of the RhoA-associated kinase ROCK1/2 restored a normal phenotype and proplatelet formation. Overall, this work suggests a new etiology for macrothrombocytopenia, in which increased RhoA activity is associated with disrupted FLNa/αIIbβ3 interaction.
Introduction
Filamins form a family of actin-binding proteins, mainly involved in the organization of the 3-dimensional F-actin filament network. There are 3 isoforms (FLNa, FLNb, and FLNc), encoded by 3 genes (FLNA, FLNB, and FLNC). FLNa is the most expressed isoform in hematopoietic cells and forms a dimer via the C-terminal domains. Interaction with the actin cytoskeleton is due principally to the actin-binding domain (ABD) at the N-terminus. The rest of the protein is composed of 24 immunoglobulin-like domains of ∼100 amino acids each, loosely organized in 2 rod-like superstructures. Like the ABD, the first rod can interact with actin, whereas the second rod is mainly responsible for the multiple interactions of FLNa with >80 different partners. Among them are integrins, cell-surface receptors essential for the regulation of cellular morphology and motility.1 Mutations in the FLNA gene are associated with a wide spectrum of rare disorders, characterized by developmental anomalies affecting multiple tissues. These disorders are generically called filaminopathies A, and the most frequent are periventricular nodular heterotopia and disorders on the otopalatodigital spectrum.2 These conditions are often associated with macrothrombocytopenia and a bleeding tendency.3 Specifically regarding megakaryocytes (MKs), the lack of FLNa induces macrothrombocytopenia; there is controversy over an associated defect in the expression of the GPIb-IX-V complex.4-6
Because the gene is localized on the X chromosome, and hemizygosity is generally considered lethal, most reported patients are women carrying heterozygous mutations. In these patients, 2 platelet populations differing in size are observed, and only a small fraction (10% to 20%) of platelets seem to lack FLNa. Moreover, no defect in MK differentiation has been observed, except for proplatelet formation.7 We previously showed that a low platelet count was associated with a reduced FLNa level compared with the control, whereas a near-normal FLNa level correlated with a normal platelet count.8 An X-inactivation skewing was excluded by analysis of T cells, leading us to hypothesize that patients’ bone marrow harbored 2 MK populations: 1 able to correctly differentiate MKs, giving rise to normal-size platelets, and the other displaying defective production of macroplatelets. This would explain in part the heterogeneity of platelet populations in filaminopathy A patients. Additionally, the large variability in platelet counts, as well as in wild-type (WT) FLNa content, suggests an anomaly at the platelet production and/or survival level.
The clonal hypothesis prompted us to develop a model based on induced pluripotent stem cells (iPSCs); this would allow the study of megakaryopoiesis, with cells expressing either the WT (FLNAWT) or mutated (FLNAmut) allele. In fact, the cellular reprogramming allowed the generation of clonal cell lines expressing only 1 X chromosome; this means it was possible to compare 2 cell lines with the same genetic background, differing only in expression of the FLNAWT or FLNAmut allele. This approach thus prevented the variability associated with the presence of 2 cellular populations in patient primary hematopoietic cells.7 MK differentiation of FLNAmut iPSCs seemed normal, but proplatelet formation was strongly impaired. Moreover, we observed abnormal stress fibers composed of F-actin bundles, correlating with the unexpected increased activity of RhoA, when cells were plated on fibrinogen but not on other extracellular matrices like collagen 1, fibronectin, or von Willebrand factor (VWF). This observation pointed to a specific role for αIIbβ3 in the regulation of RhoA and for the F-actin cytoskeleton in proplatelet formation. Accordingly, deletion of the β33 and GTPase FLNa binding regions strongly affected both proplatelet formation and RhoA activation. Overall, our observations show that FLNa downregulates RhoA activity via αIIbβ3 during proplatelet formation and that the dysregulation of the FLNa-αIIbβ3-RhoA axis leads to macrothrombocytopenia.
Materials and methods
iPSC generation and expansion
CD34+ cells were isolated from peripheral blood using an immunomagnetic bead cell-sorting system (AutoMacs; Miltenyi Biotec, Paris, France) and grown in serum-free medium containing erythropoietin (1 U/mL), FMS-like tyrosine kinase 3 ligand (FLT3L; 10 ng/mL), granulocyte colony-stimulating factor (20 ng/mL), interleukin-3 (IL-3; 10 ng/mL), IL-6 (10 ng/mL), stem cell factor (SCF; 25 ng/mL), thrombopoietin (TPO; 10 ng/mL), and granulocyte-macrophage colony-stimulating factor (10 ng/mL) for 6 days. Cells were then transduced with the CytoTune iPS 2.0 Sendai Reprogramming Kit (Thermo Fisher, Villebon-sur-Yvette, France), and reprogramming was performed according to manufacturer instructions. Colonies with an embryonic stem (ES) cell–like morphology were manually isolated, expanded for a small number of passages, and frozen. iPSCs were maintained in Essential 8 or Essential 8 Flex medium (Gibco/Thermo Fisher), on plates coated with N-truncated human recombinant vitronectin (Gibco). Cell passages were performed using a solution of 0.5 mM of EDTA in phosphate-buffered saline 1× or TrypLE 1× (Gibco). Mycoplasma screening was routinely performed, according to manufacturer instructions (Sigma, Saint-Quentin Fallavier, France). Cells were kept in culture for a limited number of passages to prevent the surge of any genomic anomalies. A list of manufacturers is provided in supplemental Table 1, available on the Blood Web site.
iPSC hematopoietic differentiation
Clumps of pluripotent cells were seeded on Geltrex-coated plates in Essential 8 medium at day −1. The starting cell concentration was adjusted for each cell line at a 10% to 20% confluency range. At day 0, cells were transferred in a xeno-free medium based on StemPro-34 SFM (Gibco), supplemented with 1% penicillin/streptomycin (volume/volume; Gibco), 1% L-glutamine (volume/volume; Gibco), 0.04 mg/mL of 1-thioglycerol (Sigma), and 50 mg/mL of ascorbic acid (Sigma). This medium was retained for the entire experiment and supplemented with different cytokines, small molecules, and growth factors, according to the following schedule: days 0 to 2: bone morphogenetic protein 4 (10 ng/mL), vascular endothelial growth factor (VEGF; 50 ng/mL), and CHIR99021 (2 μM); days 2 to 4: bone morphogenetic protein 4 (10 ng/mL), VEGF (50 ng/mL), and fibroblast growth factor 2 (FGF2; 20 ng/mL); days 4 to 6: VEGF (15 ng/mL) and FGF2 (5 ng/mL); day 6: VEGF (50 ng/mL), FGF2 (50 ng/mL), SCF (50 ng/mL), and FLT3L (5 ng/mL); days 7 to 10: VEGF (50 ng/mL), FGF2 (50 ng/mL), SCF (50 ng/mL), FLT3L (5 ng/mL), TPO (50 ng/mL), and IL-6 (10 ng/mL); and days 10 to 20: SCF (50 ng/mL), FLT3L (5 ng/mL), TPO (50 ng/mL), and IL-6 (10 ng/mL). A list of manufacturers is provided in supplemental Table 1.
Immunofluorescence and confocal imaging
Slides were coated with 25 μg/mL of fibrinogen (Sigma) for 2 hours at room temperature. Purified MKs were plated on fibrinogen for 1 hour at 37°C. Cells were fixed with 4% paraformaldehyde for 10 minutes and permeabilized with a buffer containing 0.2% Triton X-100, 100 mM of PIPES (pH, 6.9), 2 M of glycerol, 1 mM of EGTA, and 1 mM of magnesium chloride for 5 minutes. Primary and secondary antibodies were diluted in PBS containing 0.1% bovine serum albumin (weight/volume); incubation was performed for 1 hour for each antibody. Concentration, clone name, and manufacturer for each antibody are listed in supplemental Table 2. Slides were mounted using Vectashield with DAPI (Molecular Probes/Thermo Fisher). Proximity ligation assay (Sigma) was performed according to manufacturer instructions. Images were acquired using a Leica DMI 4000 SPE laser-scanning microscope with a 63×/1.4 numeric aperture oil objective (Leica Microsystem, Lognes, France). Stress fibers were scored on a minimum of 100 cells per slide. Image analysis was performed with LASX software (Leica Microsystem).
Proplatelet formation assay
MKs isolated at day 14 of culture were seeded at a cell density of 5 × 103 cells per well in a 96-well plate in serum-free medium containing TPO (10 ng/mL) and SCF (25 ng/mL). Proplatelet-forming cells were scored after 4 to 5 days by enumeration of ≥200 cells per well using an inverted microscope (Carl Zeiss, Marly-le-Roi, France) at a 200× magnification. A proplatelet-forming MK was defined as a cell displaying ≥1 cytoplasmic process with a clearly defined constriction area. Each condition was examined in triplicate.
FRET analysis
The lentiviral RhoA fluorescence resonance energy transfer (FRET) sensor (pPBbsr2-Raichu-2707×) was a kind gift from Dr. Matsuda. Undifferentiated iPSCs were infected and selected for expression of CFP and YPet reporters by fluorescence-activated cell sorting. iPS-derived MKs were isolated by fluorescence-activated cell sorting and plated on fibrinogen (25 μg/mL), collagen 1 (50 μg/mL), fibronectin (25 μg/mL), N-truncated vitronectin (50 μg/mL), or VWF (25 μg/mL) for 2 hours at 37°C. Slides were mounted with Fluoromount-G (Southern Biotech/Clinisciences, Nanterre, France). FRET was performed on a confocal microscope Leica SP8 with a 63× objective (1.4 NA) with the acceptor photobleaching method using FRET-AB Wizard in Leica software. Briefly, the acceptor fluorophore (YPet) fluorescence was bleached at 100% laser intensity, ensuring a minimal extent of bleaching by 75% and resulting in an increased fluorescence intensity of the donor fluorophore (CFP) in the bleached region, which was subsequently measured. FRET efficiency was obtained using the formula:
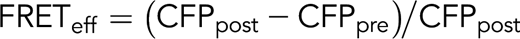
Statistics
All data are shown as mean ± standard deviation unless specified differently in the legend. Statistical analyses were performed using PRISM software (GraphPad software). Statistical significance was established using a Student t test as specified in legends or 1-way analysis of variance followed by all pairwise multiple comparison procedures (Student-Newman-Keuls method). Differences were considered significant at P < .05.
Study approval
The study was performed under the promotion of the ethics committee of Ile de France IV (IRB 00003835) and in accordance with the Declaration of Helsinki. All participants provided informed consent before participation in this study.
Other materials and methods could be found in the data supplement.
Results
Patient mutant iPSCs display unstable FLNA mRNA and no FLNa
iPSC clones were derived from 2 female patients, P1 (238 × 109 platelets per L) and P2 (110 × 109 platelets per L), characterized by deletion of exons 31 to 48 (P1) and 20 to 48 (P2), respectively (patients P4 and P2 in our initial study8 ). Cellular reprogramming allowed the generation of clonal cell lines that stably expressed the X chromosome carrying either the FLNAWT or the FLNAmut allele. Therefore, a panel of isogenic lines could be issued from the same genetic background, allowing direct comparison. FLNAWT and FLNAmut clones were screened for each patient, using 2 quantitative reverse transcription polymerase chain reactions able to discriminate between the 2 alleles (Figure 1A). The screening did not show any evident difference in the percentage of generated FLNAmut clones compared with the control; nevertheless, the FLNA mRNA level was significantly decreased by ≥50% in all FLNAmut clones compared with FLNAWT clones (Figure 1B), suggesting early instability of the mRNA. WT FLNa was detected in FLNAWT clones; however, no truncated forms of FLNa protein were detected in FLNAmut clones (Figure 1C-D). Putative XCI erosion9 affecting the FLNA locus was ruled out by immunoblot after maintaining the iPSC clones in culture for 60 passages (Figure 1E; supplemental Figure 1).
Patient iPSCs display unstable FLNA mRNA and no FLNa. (A) Quantitative reverse transcription polymerase chain reaction (qRT-PCR) primer design for the identification of the expressed X chromosome. Amplification of exon 2 allows detection of both WT and mutated messenger RNA (mRNA); that of exon 43 allows detection of only the WT mRNA. (B) qRT-PCR for FLNa expression on 2 different exons for several iPSC clones (9 FLNAWT and 20 FLNAmut clones for P1; 9 FLNAWT and 10 FLNAmut clones for P2). Results are presented as mean ± standard error of the mean; unpaired Student t test with Welch’s correction was used; each point represents 1 independent experiment. (C-D) Representative immunoblots for FLNa expression on 4 iPSC clones for each patient using a C-terminus–specific antibody (C) and a N-terminal antibody (D). Lines 1, 3, 5, and 7 represent FLNAWT clones, and lines 2, 4, 6, and 8 represent FLNAmut clones. (E) Immunoblot for FLNa expression on iPSCs cultivated for 60 passages using C-terminal antibody. Line 1 represents FLNAWT clone, and lines 2 to 4 represent 3 different FLNAmut clones. ***P < .001.
Patient iPSCs display unstable FLNA mRNA and no FLNa. (A) Quantitative reverse transcription polymerase chain reaction (qRT-PCR) primer design for the identification of the expressed X chromosome. Amplification of exon 2 allows detection of both WT and mutated messenger RNA (mRNA); that of exon 43 allows detection of only the WT mRNA. (B) qRT-PCR for FLNa expression on 2 different exons for several iPSC clones (9 FLNAWT and 20 FLNAmut clones for P1; 9 FLNAWT and 10 FLNAmut clones for P2). Results are presented as mean ± standard error of the mean; unpaired Student t test with Welch’s correction was used; each point represents 1 independent experiment. (C-D) Representative immunoblots for FLNa expression on 4 iPSC clones for each patient using a C-terminus–specific antibody (C) and a N-terminal antibody (D). Lines 1, 3, 5, and 7 represent FLNAWT clones, and lines 2, 4, 6, and 8 represent FLNAmut clones. (E) Immunoblot for FLNa expression on iPSCs cultivated for 60 passages using C-terminal antibody. Line 1 represents FLNAWT clone, and lines 2 to 4 represent 3 different FLNAmut clones. ***P < .001.
FLNa deficiency induces marked defect in proplatelet formation
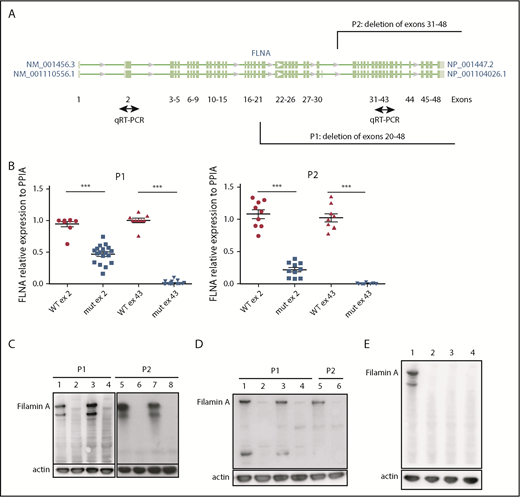
Two FLNAWT and 2 FLNAmut clones for each patient were selected for further study and validated for stable karyotype and functional pluripotency (supplemental Figure 2). MK differentiation was induced using an updated version of the protocol described by Chou et al10 (supplemental Figure 3). Absence of FLNa was confirmed by immunofluorescence of mature CD41+CD42+ MKs (Figure 2A). Surprisingly, an abnormal F-actin distribution in large stress fibers was observed in FLNAmut MKs, suggesting GTPase RhoA activation. Quantification by flow cytometry of MKs derived from FLNAWT and FLNmut iPSCs showed similar percentages of CD41+CD42+ cells (Figure 2Bi-ii), and no difference in mean fluorescence intensity was detected for αIIbβ3 (anti-CD41a), GPIX (anti-CD42a), or GPIbα (anti-CD42b) expression (Figure 2Biii). This suggested that both αIIbβ3 and GPIb-IX-V were normally expressed at the cell surface of mature MKs. A marked decrease in the percentage of proplatelet-forming MKs was observed in FLNAmut clones for both patients (Figure 2C), consistent with the previous observation in CD34+-derived MKs from patient P2.7 Moreover, the FLNAmut MKs displayed proplatelets with platelet-like tips larger than those in FLNAWT clones (Figure 2D). The proplatelet formation defect was not due to an accelerated MK maturation, because the differential pattern between FLNAWT and FLNAmut MKs remained unchanged throughout culture. To rule out a possible partial compensation effect by the FLNB isoform, we measured the expression of the 2 genes during megakaryopoiesis (supplemental Figure 4A). We noted a strong upregulation of FLNA mRNA in the late stages of megakaryopoiesis, but no FLNB mRNA upregulation was detected in the absence of FLNa (supplemental Figure 4B), suggesting that FLNb has no compensatory effect. We conclude that our model recapitulates the main features observed in the patients and that it is suitable for addressing the mechanism of the defective proplatelet formation.
FLNa deficiency induces a marked defect in proplatelet formation. (A) Immunofluorescence staining for the expression of FLNa (green), F-actin (red), and nucleus (blue) in iPSC-derived MKs from P1; scale bar = 30 μm. (B) Flow cytometry analysis of CD41a, CD42a, and CD42b expression: representative dot plot (i) and relative percentages (ii) (n = 10); histogram representative of mean fluorescence intensity for CD41a and CD42a (n = 5) and for CD42b (n = 3); paired Student t test (iii). (C) Proplatelet formation potential of patient iPSC-derived clones. At least 2 clones were assayed for each genotype, for each patient, at 2 different time points. Results are presented as mean ± standard error of the mean; unpaired Student t test with Welch’s correction was used; each point represents 1 independent experiment. (A-B) Experiments were performed at day 15 or 16 of MK differentiation. (C) Proplatelet formation was measured at days 18 and 19. (D) Representative pictures for WT and mutant proplatelet-forming MKs from P1. Immunofluorescence staining of F-actin (green) and β-tubulin (red) was performed after adhesion on fibrinogen for 24 hours. The nucleus is stained with 4′,6-diamidino-2-phenylindole (DAPI; blue); scale bar = 30 μm. *P < .05, **P < .01, ***P < .001.
FLNa deficiency induces a marked defect in proplatelet formation. (A) Immunofluorescence staining for the expression of FLNa (green), F-actin (red), and nucleus (blue) in iPSC-derived MKs from P1; scale bar = 30 μm. (B) Flow cytometry analysis of CD41a, CD42a, and CD42b expression: representative dot plot (i) and relative percentages (ii) (n = 10); histogram representative of mean fluorescence intensity for CD41a and CD42a (n = 5) and for CD42b (n = 3); paired Student t test (iii). (C) Proplatelet formation potential of patient iPSC-derived clones. At least 2 clones were assayed for each genotype, for each patient, at 2 different time points. Results are presented as mean ± standard error of the mean; unpaired Student t test with Welch’s correction was used; each point represents 1 independent experiment. (A-B) Experiments were performed at day 15 or 16 of MK differentiation. (C) Proplatelet formation was measured at days 18 and 19. (D) Representative pictures for WT and mutant proplatelet-forming MKs from P1. Immunofluorescence staining of F-actin (green) and β-tubulin (red) was performed after adhesion on fibrinogen for 24 hours. The nucleus is stained with 4′,6-diamidino-2-phenylindole (DAPI; blue); scale bar = 30 μm. *P < .05, **P < .01, ***P < .001.
αIIbβ3 and Rho GTPase FLNa interaction domains are essential for proplatelet formation
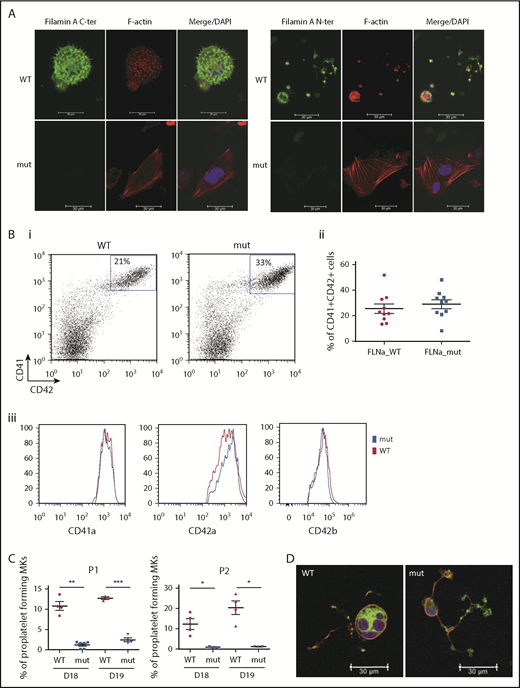
Taking advantage of the absence of FLNa in the FLNAmut clones, we decided to transduce cells from the P1 FLNAmut iPSC clone with several FLNa complementary DNAs (cDNAs) lacking domains essential for MK maturation: a mutant lacking the ABD at the N-terminus (del1),11 a mutant lacking the Ig17 domain that interacts with GPIbα (del2),12 a mutant lacking the Ig21 domain that interacts with the β3 integrin (del3),13 and a mutant lacking the dimerization domain involved in the interaction with small GTPases (del4).14 The full-length FLNA cDNA (mutant + WT) was also introduced as an internal control (Figure 3A). For all these constructs, we used a zinc finger nuclease–based technology targeting the AAVS1 locus as previously described.15 Expression and relative stability of the mutant FLNa proteins were verified in all newly generated iPSC clones (Figure 3B). Moreover, the absence of interaction of FLNa with GPIbα, β3 integrin, or RhoA GTPase using the del2, del3, or del4 mutant, respectively, was checked in iPSC-derived MKs by proximity ligation assay (supplemental Figure 5; Figure 3C). Regarding MK differentiation, no difference in the percentage of CD41+CD42+ MKs was observed (Figure 3D). Mutants lacking the domains interacting with actin (del1), β3 integrin (del3), and Rho GTPases (del4) displayed reduced proplatelet formation potential. These results are consistent with a central role for the interaction of FLNa with αIIbβ3, RhoA, and actin in proplatelet formation. Of note, del2 mutant (unable to interact with GPIbα) yielded proplatelet-forming MKs in numbers similar to FLNAWT and FLNAmut+WT MKs (Figure 3E). These results clearly show that the interactions of FLNa with β3 integrin and RhoA GTPase are essential for proplatelet formation.
Deletion of αIIbβ3and Rho GTPase FLNa interaction domains, but not of GPIbα-interacting domain, deeply affects proplatelet formation. (A) Schematic representation of FLNa mutants introduced by zinc finger nuclease–mediated gene editing. (B) Representative immunoblot for FLNa expression in the iPSC-edited clones using an N-terminus (left) and C-terminus (right) antibody. (C) Representative pictures of proximity ligation assay for FLNa/β3 and FLNa/RhoA interactions. Red staining represents the interaction between β3 and FLNa and between RhoA and FLNa. The nucleus is stained with 49,6-diamidino-2-phenylindole (blue); scale bar = 30 μm. Two independent experiments for each interaction were performed, with 50 cells analyzed in each experiment. No specific signal was detected when cells were incubated without primary antibodies (data not shown). (D) Flow cytometry analysis of CD41a and CD42a expression in the edited clones at day 15 of differentiation. Results are presented as mean ± standard error of the mean; unpaired Student t test with Welch’s correction was used; each point represents 1 independent experiment. (E) Proplatelet formation potential evaluated at day 18 of MK differentiation for each edited clone (n = 3). Paired Student t test *P < .05, **P < .01.
Deletion of αIIbβ3and Rho GTPase FLNa interaction domains, but not of GPIbα-interacting domain, deeply affects proplatelet formation. (A) Schematic representation of FLNa mutants introduced by zinc finger nuclease–mediated gene editing. (B) Representative immunoblot for FLNa expression in the iPSC-edited clones using an N-terminus (left) and C-terminus (right) antibody. (C) Representative pictures of proximity ligation assay for FLNa/β3 and FLNa/RhoA interactions. Red staining represents the interaction between β3 and FLNa and between RhoA and FLNa. The nucleus is stained with 49,6-diamidino-2-phenylindole (blue); scale bar = 30 μm. Two independent experiments for each interaction were performed, with 50 cells analyzed in each experiment. No specific signal was detected when cells were incubated without primary antibodies (data not shown). (D) Flow cytometry analysis of CD41a and CD42a expression in the edited clones at day 15 of differentiation. Results are presented as mean ± standard error of the mean; unpaired Student t test with Welch’s correction was used; each point represents 1 independent experiment. (E) Proplatelet formation potential evaluated at day 18 of MK differentiation for each edited clone (n = 3). Paired Student t test *P < .05, **P < .01.
Loss of FLNa/αIIbβ3 interaction increases RhoA activity
After adhesion of iPSC-derived FLNAmut MKs on fibrinogen (Figure 2A), an increase in stress fiber formation in FLNAmut clones was observed. In light of this unexpected observation, we investigated in more detail the stress fiber formation on fibrinogen matrix by different mutant MKs. A majority of FLNAmut MKs displayed a large number of stress fibers compared with the FLNAWT and FLNAmut+WT clones (Figure 4A-B). Stress fibers were also markedly enhanced in the mutants lacking either the β3 integrin–interacting domain (del3) or the dimerization domain involved in the interaction with small GTPases (del4; Figure 4B). As opposed to fibrinogen, no difference in stress fiber formation was detected on fibronectin, which interacts with αIIbβ3 with a much lower affinity (supplemental Figure 6). To confirm the role of the RhoA pathway in cells lacking FLNa, we directly compared RhoA activity in cells settled on fibrinogen (αIIbβ3 ligand), fibronectin (α5β1 ligand), vitronectin (αVβ3 ligand), collagen 1 (αIIβ1 ligand), and VWF (GPIb-GPIX-GPV ligand; Figure 4C) using a lentiviral FRET biosensor based on the Raichu probe.16 On fibrinogen, the absence of FLNa led to a threefold increase in RhoA activity compared with the control, which was significantly reduced when FLNa full-length cDNA in FLNAmut MKs was expressed (Figure 4Cii). Moreover, treatment of MKs with a catalytic inhibitor of ROCK1/2 (Y-27632), a major effector of RhoA-mediated signaling in MKs, had no effect on RhoA activity, supporting the hypothesis of a direct alteration of RhoA activity (supplemental Figure 7). In contrast, no significant difference in RhoA activity was observed in FLNAWT or FLNAmut MKs settled on other substrates (fibronectin, vitronectin, collagen 1, and VWF).
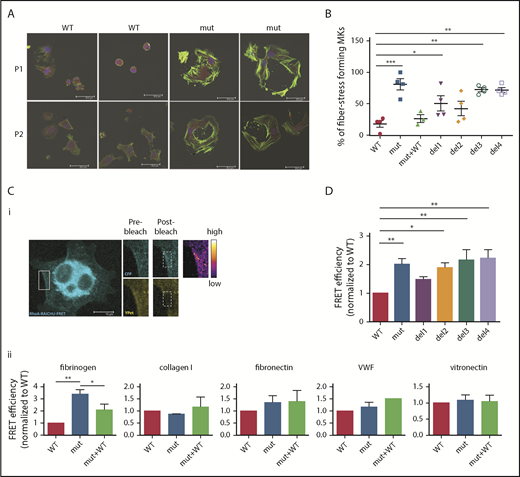
RhoA is overactivated in FLNa-deficient MKs. (A) Immunofluorescence staining of G-actin (red) and F-actin (green) after adhesion on fibrinogen. The nucleus is stained with 4′,6-diamidino-2-phenylindole (blue); scale bar = 30 μm. (B) Quantification of the percentage of stress fiber–forming MKs for each edited clone. Results are presented as mean ± standard error of the mean; each point represents 1 independent experiment. Data were analyzed by performing 1-way analysis of variance (ANOVA) followed by all pairwise multiple comparison procedures (Student-Newman-Keuls method). (C) FRET analysis for RhoA activation on different substrates: representative image, scale bar = 10 μm (i) and adhesion on fibrinogen (n = 4), collagen 1 (n = 3), fibronectin (n = 6), VWF (n = 3), and vitronectin (n = 5) (ii). (D) FRET analysis of RhoA activation on fibrinogen of all edited clones: WT and mutant clones (n = 8), del1 and del4 clones (n = 6), and del2 and del3 clones (n = 5). (C-D) At least 15 cells per condition were analyzed. Data were analyzed by performing ANOVA followed by all pairwise multiple comparison procedures (Student-Newman-Keuls method); all experiments were performed at day 15 or 16 of MK differentiation. *P < .05, **P < .01, ***P < .001.
RhoA is overactivated in FLNa-deficient MKs. (A) Immunofluorescence staining of G-actin (red) and F-actin (green) after adhesion on fibrinogen. The nucleus is stained with 4′,6-diamidino-2-phenylindole (blue); scale bar = 30 μm. (B) Quantification of the percentage of stress fiber–forming MKs for each edited clone. Results are presented as mean ± standard error of the mean; each point represents 1 independent experiment. Data were analyzed by performing 1-way analysis of variance (ANOVA) followed by all pairwise multiple comparison procedures (Student-Newman-Keuls method). (C) FRET analysis for RhoA activation on different substrates: representative image, scale bar = 10 μm (i) and adhesion on fibrinogen (n = 4), collagen 1 (n = 3), fibronectin (n = 6), VWF (n = 3), and vitronectin (n = 5) (ii). (D) FRET analysis of RhoA activation on fibrinogen of all edited clones: WT and mutant clones (n = 8), del1 and del4 clones (n = 6), and del2 and del3 clones (n = 5). (C-D) At least 15 cells per condition were analyzed. Data were analyzed by performing ANOVA followed by all pairwise multiple comparison procedures (Student-Newman-Keuls method); all experiments were performed at day 15 or 16 of MK differentiation. *P < .05, **P < .01, ***P < .001.
Finally, RhoA activity was measured in all mutants (del1 to del4) on fibrinogen matrix. A significant increase in Rho activity was observed for the mutants lacking interaction with β3 (del3) and interaction with RhoA (del4). RhoA activity was also increased with the mutant lacking the GPIbα interaction domain (del2), but to a lesser extent than del3 and del4 (Figure 4D). If combined with the lack of any significant difference in stress fiber formation (Figure 4B) or proplatelet formation (Figure 3E), these data together suggest that in our model, FLNa-GPIbα is not significantly involved in proplatelet formation, whereas the αIIbβ3-FLNa-RhoA axis seems to play a more important role.
RhoA activity inhibition prevents stress fiber formation and restores proplatelet development
To confirm the involvement of the RhoA/ROCK pathway, FLNAWT and FLNAmut MKs were treated with a ROCK1/2 inhibitor (Y-27632) and then assessed for stress fiber formation (Figure 5A) and proplatelet development (Figure 5B-C). A significant reduction in the number of stress fiber–forming MKs was observed for all conditions, but to a greater extent for FLNAmut, del3, and del4 clones (Figure 5A; supplemental Figure 8).
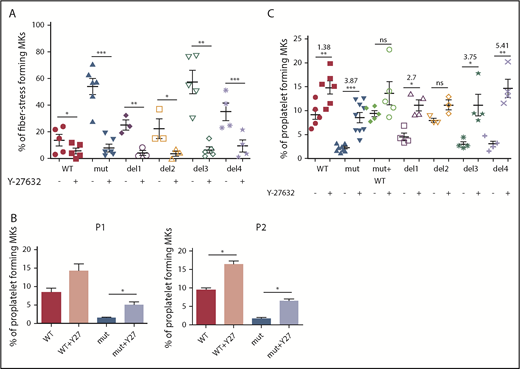
Proplatelet development and stress fiber formation are rescued after inhibition of RhoA pathway. (A) Stress fiber formation assessed at day 15 or 16 of MK differentiation in presence and absence of ROCK1/2 inhibitor Y-27632 for the edited clones: WT and del3 (n = 5), mutant (n = 6), del1 and del2 (n = 3), and del4 (n = 4); paired Student t test. (B) Proplatelet formation potential in presence and absence of ROCK1/2 inhibitor Y-27632 for both patients’ iPSC clones (n = 3); paired Student t test. (C) Proplatelet formation potential in presence and absence of ROCK1/2 inhibitor Y-27632 in all edited clones: WT (n = 6), mutant (n = 8), mutant + WT, del1, del3, and del4 (n = 4), and del2 (n = 3); paired Student t test. The ROCK1 inhibitor was added at day 15 to culture, and proplatelet formation was measured at day 18. *P < .05, **P < .01, ***P < .001.
Proplatelet development and stress fiber formation are rescued after inhibition of RhoA pathway. (A) Stress fiber formation assessed at day 15 or 16 of MK differentiation in presence and absence of ROCK1/2 inhibitor Y-27632 for the edited clones: WT and del3 (n = 5), mutant (n = 6), del1 and del2 (n = 3), and del4 (n = 4); paired Student t test. (B) Proplatelet formation potential in presence and absence of ROCK1/2 inhibitor Y-27632 for both patients’ iPSC clones (n = 3); paired Student t test. (C) Proplatelet formation potential in presence and absence of ROCK1/2 inhibitor Y-27632 in all edited clones: WT (n = 6), mutant (n = 8), mutant + WT, del1, del3, and del4 (n = 4), and del2 (n = 3); paired Student t test. The ROCK1 inhibitor was added at day 15 to culture, and proplatelet formation was measured at day 18. *P < .05, **P < .01, ***P < .001.
Finally, proplatelet formation was assessed for both patient-derived iPSC clones in the presence or absence of a ROCK1/2 inhibitor. As shown in Figure 5B, in the absence of FLNa and presence of a ROCK1/2 inhibitor, proplatelet formation was partially restored, confirming that RhoA downregulation is crucial for proplatelet formation. Finally, we tested the proplatelet formation potential in the presence of a ROCK1/2 inhibitor for all edited mutants (del1 to del4), and we observed a higher increase for del3 (×3.75) and del4 (×5.41) than for del1 (×2.7) and WT (×1.38), confirming once again the importance of the αIIbβ3-FLNa-RhoA axis for proplatelet formation (Figure 5C). In conclusion, these results show that ROCK1/2 inhibition restores a normal phenotype in the absence of FLNa.
Discussion
FLNa is a central actor in linking the cytoskeleton to several receptors, allowing signal transduction after adhesion to the extracellular matrix. Mutations in the FLNA gene are associated with a wide spectrum of rare diseases,3 sometimes associated with macrothrombocytopenia and a bleeding tendency.7 Several studies have investigated the precise role of FLNa in megakaryopoiesis. In mice, although the homozygous Flna deletion was embryonically lethal,2 heterozygous Flna deletion in females led to mild thrombocytopenia, with >95% of platelets of normal size expressing FlnA and only 5% of larger platelets lacking FlnA.4 The Flna knockout mouse model, specifically for the erythromegakaryocytic lineage, resulted in severe macrothrombocytopenia with increased tail bleeding time.4 In this model, platelets displayed an altered cytoskeleton and linkage of GPIbα to actin filaments, associated with a decreased level of GPIb-GPIX-GPV.4 The MK-specific Flna knockout model showed a premature release of large and fragile platelets, and thrombocytopenia was attributed to increased platelet clearance, mediated by macrophages. Although FlnAnull MKs displayed normal expression of all VWF receptor subunits, their surface expression was reduced in platelets.5 In the last model, a decreased surface expression of GPIbα was detected in MKs derived from murine ES cells where Flna and Flnb expression was decreased via short hairpin RNA inhibition. MK differentiation was altered, the proplatelets exhibited a distinctly abnormal morphology with enlarged swellings and thick shafts, and the platelets were 2 to 3 times larger than control proplatelets/platelets. An equilibrated stoichiometry of FLNa and GPIbα was shown to be necessary for optimal trafficking to the margin of the cell and controlling the platelet size in an overexpression-based cellular model.6 These conflicting reports, combined with difficulties in the human modeling of this disorder, have not clearly addressed the macrothrombocytopenia etiology at the megakaryocytic level.
Using a patient-specific model, we demonstrated here the total absence of FLNa in the iPSCs and MKs expressing FLNAmut. This could be explained by the fact that deletions may correspond to regions responsible for the stabilization of the protein, such as the phosphorylation of Ser2152 (exon 39, included within the region deleted in both patients), which allows protection against proteolysis.17 This may be related to the severe macrothrombocytopenia caused by mutations in the catalytic domain of PKA, the kinase for S2152 phosphorylation.18 No defects in MK differentiation or in CD42a (GPIX), CD42b (GPIbα), or CD41a (GPIIb) surface expression in FLNAmut iPSCs were observed. These results are in contrast to the defective MK differentiation reported for mouse ES cells with decreased Flna expression,6 but they are consistent with the data reported in mice5 and confirm that in human megakaryopoiesis the expression of the GPIb-IX-V complex is normal in the absence of FLNa. However, despite normal GPIb-IX-V surface expression, we detected a major defect in proplatelet formation, in agreement with the observation of Nurden et al.7 To understand the etiology of this defect, we investigated the role of some of the most important molecular partners of FLNa in megakaryopoiesis and thrombopoiesis by overexpression of domain-deleted FLNa constructs in FLNAmut cells. Although the impaired proplatelet formation in the FLNa mutant lacking the actin interaction (del1) was not surprising, an almost normal formation of proplatelets in the mutant lacking the interaction with GPIbα (del2) was unexpected. Indeed, GPIbα is a causal gene for macrothrombocytopenia in Bernard-Soulier syndrome,19 and the defect in its surface expression leads to defective proplatelet formation.20,21 Moreover, recent evidence suggests that GPIb plays a central role in transendothelial proplatelet biogenesis through the regulation of the GPIb-CDC42-RhoA axis.22 The role of GPIbα/FLNa interaction in resistance to shear during platelet adhesion23 and the importance of shear in proplatelet production24 suggest that this interaction is also important for proplatelet formation. Our data show that the disruption of GPIbα/FLNa interaction does not directly alter proplatelet formation in static conditions. However, these results do not exclude a role for this interaction in flow conditions, such as in transendothelial platelet biogenesis.
In contrast, overexpression of the mutant lacking the domain interacting with small GTPases (del4) led to a deep defect in proplatelet formation, confirming the importance of the interaction between RhoA and FLNa in this process. It is known that abnormal RhoA activity inhibits proplatelet formation and that RhoA activity is physiologically downregulated during the latest phases of megakaryopoiesis.25,26 FLNa absence was shown to lead to an increase in RhoA activity in other cell types,27 but this is the first report demonstrating a direct interaction between RhoA and FLNa in MKs. FLNa is also known to interact with different GAPs and GEFs28 ; therefore, it would be interesting to identify which of these partners is able to modulate RhoA activity in the absence of FLNa in MKs.
Unexpectedly, an increase in stress fiber–forming MKs lacking FLNa upon adhesion on fibrinogen was observed. Under normal conditions, RhoA-dependent stress fiber formation is triggered by adhesion to collagen 125 but not to fibrinogen, the main ligand for the integrin αIIbβ3. The use of the mutant lacking the domain interacting with β3 integrin (del3) clearly shows the crucial role of αIIbβ3/FLNa interaction in maintaining the RhoA pathway in an inactive state. These results were reinforced by the fact that the increased RhoA activity was only observed on fibrinogen but not on other matrices, such as collagen 1, fibronectin, vitronectin, or VWF. The increased in vitro proplatelet formation on fibrinogen compared with other matrices has been reported previously for mouse MKs,29 demonstrating the central role of αIIbβ3 for proplatelet formation on fibrinogen. Of note, adhesion on fibrinogen was also shown to regulate proplatelet formation in human MKs.30 Together with the fact that fibrinogen is localized next to vascular sinusoids,29 these results clearly show an active role of fibrinogen-dependent signaling in proplatelet formation and platelet release.
It is possible that αIIbβ3-dependent RhoA activation could be involved in other macrothrombocytopenias; for example, this newly described mechanism could be responsible for macrothrombocytopenia observed in thrombasthenic patients with specific αIIbβ3 mutations.31,32 Moreover, this new mechanism should be considered in patient stratification, because we observed that the chemical inhibition of RhoA signaling exhibited a positive effect on proplatelet formation in the absence of FLNa. Furthermore, ROCK1/2 inhibition increased the number of platelets generated in an ex vivo system, supporting this potential therapeutic strategy.33
Overall, these results clearly demonstrate that the absence of FLNa in MKs leads to an αIIbβ3-dependent overactivation of the RhoA pathway, which seems to be a predominant actor responsible for the observed deep defect in proplatelet formation (Figure 6). Whether RhoA activation is involved in the increased size of platelets in the absence of FLNa remains to be investigated.
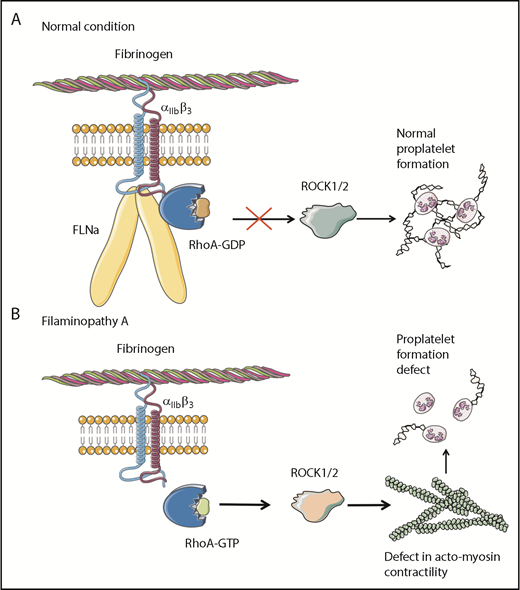
Pathological mechanism for FLNa-deficient MKs. (A) In the presence of FLNa, the interaction between fibrinogen and its receptor αIIbβ3 does not trigger RhoA pathway activation. No anomalies in proplatelet formation could be observed. (B) In the absence of FLNa, the interaction between fibrinogen and αIIbβ3 leads to an increase in RhoA activity. Consequently, the normal actomyosin contractility is disrupted via ROCK1/2 activity, and this leads to deeply flawed proplatelet formation. This increased RhoA activity in the absence of FLNa is specifically dependent on fibrinogen and absent in the presence of other extracellular matrices like fibronectin, vitronectin, collagen 1, or VWF.
Pathological mechanism for FLNa-deficient MKs. (A) In the presence of FLNa, the interaction between fibrinogen and its receptor αIIbβ3 does not trigger RhoA pathway activation. No anomalies in proplatelet formation could be observed. (B) In the absence of FLNa, the interaction between fibrinogen and αIIbβ3 leads to an increase in RhoA activity. Consequently, the normal actomyosin contractility is disrupted via ROCK1/2 activity, and this leads to deeply flawed proplatelet formation. This increased RhoA activity in the absence of FLNa is specifically dependent on fibrinogen and absent in the presence of other extracellular matrices like fibronectin, vitronectin, collagen 1, or VWF.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank the patients for participation in this study; O. Bawa and P. Opolon from the preclinical research platform, Gustave Roussy, Villejuif, France, for teratoma analysis; and P. Rameau, C. Catelain, and Y. Lecluse from the imaging and cytometry platform (PFIC), UMS AMMICA, Gustave Roussy, Villejuif, France, for expertise in cytometry.
This work was supported by French grants from Fondation pour la Recherche Medicale (LPC20170637458) and Ligue Nationale Contre le Cancer (équipe labellisée 2016; H.R.) and by European grants ERA-NET (2013; C. Balduini) and H2020-FETOPEN-1-2016-2017-SilkFusion. A.D. was supported by a PhD fellowship from the Sorbonne Paris Cité and Ligue National Contre le Cancer and H2020-FETOPEN-1-2016-2017-SilkFusion. D.S. was supported by a fellowship from ANR and Ligue National Contre le Cancer.
Authorship
Contribution: A.D., N.B., D.S., V.C., L.L., F.B., L.T., G.T., and N.D. performed and analyzed experiments; A.D., W.V., I.P., N.D., J.-P.R., M.B., and H.R. discussed results; C.G. and R.F. provided clinical and biological follow-up of patients; C.V.D. provided the biological material; H.R. supervised the work; and A.D., J.-P.R., M.B., and H.R. wrote the article.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Hana Raslova, INSERM UMR1170, Gustave Roussy Cancer Campus, 114 rue Edouard Vaillant, 94805, Villejuif cedex, France; e-mail: hana.raslova@gustaveroussy.fr.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal