

In this issue of Blood, use an isogenic human induced pluripotent stem cell (iPSC) model to show that the loss of αIIbβ3-filamin A interactions leads to RhoA activation, proplatelet formation defects, and macrothrombocytopenia.1
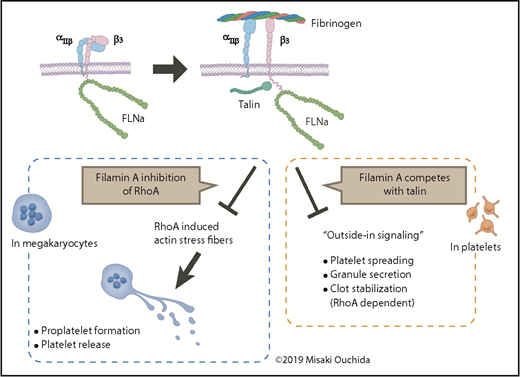
In MKs, the binding of αIIbβ3 to fibrinogen promotes PPF and platelet release. In platelets, the binding triggers outside-in signaling, partly through talin, to induce platelet spreading, granule secretion, and RhoA-dependent clot stabilization. Filamin A modulates these processes by inhibiting RhoA activity and by competing with talin.
In MKs, the binding of αIIbβ3 to fibrinogen promotes PPF and platelet release. In platelets, the binding triggers outside-in signaling, partly through talin, to induce platelet spreading, granule secretion, and RhoA-dependent clot stabilization. Filamin A modulates these processes by inhibiting RhoA activity and by competing with talin.
Human iPSCs have emerged as a powerful tool not only to regenerate human cells and tissues, but also to recapitulate disorders in vitro to identify the pathogenesis and novel therapeutic approaches. Donada and colleagues have used iPSCs to probe a congenital macrothrombocytopenia caused by mutations in the filamin A gene on the X chromosome. Because of intrinsic X chromosome inactivation, the establishment of human iPSCs resulted in 2 groups of clones silenced for either one of the X chromosomes. In other words, virtually isogenic iPSC clones expressing either mutated or wild-type genes were prepared.
Mutations in genes related to megakaryocyte (MK)-platelet cytoskeletal organization have been known to cause congenital macrothrombocytopenia, an array of disorders that present with a reduced platelet count and platelets of large to giant size in the blood circulation.2 Defects in proplatelet formation (PPF) by improper cytoskeletal rearrangement are responsible. Affected genes include β1-tubulin (TUBB1), NMMHC-IIA (MYH9), actin filament cross-linking α-actinin-1 (ACTN1), integrin αIIbβ3 (ITGA2B and ITGB3), vWF-GPIb/IX/V (VWF, GP1BA, GP1BB, and GP9), filamin A (FLNA), and filamin A-phosphorylating kinase PRKACG (PRKACG).2
Filamin A is known to connect GPIb and αIIbβ3 to actin fibers in the cytoplasm of MKs and platelets. Female patients with a heterogeneous mutation in FLNA have been shown to have thrombocytopenia along with other malformations.2,3 The circulating platelets show major and minor populations of normal-size platelets with a normal expression level of filamin A and large-size platelets with a low expression level of filamin A, respectively. These observations suggest that the defective platelets come from MKs in which the X chromosome with wild-type filamin A is silenced.
To gain insights into the mechanism of this congenital macrothrombocytopenia, mouse knockout models and mouse embryonic stem cells (ESCs) manipulated for the expression of FLNA have been studied.4,5 The macrothrombocytopenia phenotype and defects in PPF were observed in both models. However, the ESC model also showed inefficient MK differentiation, whereas knockout mice showed increased MKs in the bone marrow and spleen. Aside from the use of mouse models, the disagreement emphasizes the need for human cells to study the pathogenesis in human patients. However, studies on patient bone marrow MKs are lacking, presumably because of the invasiveness of acquiring primary cells.
Donada et al established iPSCs from 2 female patients with different filamin A mutations.1 Multiple iPSC clones expressing either wild-type or mutant filamin A were obtained, thus enabling comparisons of the phenotype in a virtually isogenic human iPSC model. Although other X chromosome genes differently expressed between the 2 groups may affect megakaryothrombopoiesis, this concern was negligible here because the phenotype observed in the mutant clones was common in the derived MKs.
First, the expression of mutant filamin A was significantly reduced, which is in accordance with a missing region that protects filamin A against proteolysis. Second, the mutant MKs showed defective PPF, whereas MK differentiation showed no apparent difference, as seen in mouse models. Interestingly, in MKs cultured on fibrinogen, an extraordinary development of F-actin fiber bundles was observed as the overactivation of RhoA, a GTPase that stimulates actin polymerization.6 Third, noting that no filamin A was expressed in the MKs derived from the iPSCs of 1 patient, the researchers overexpressed a series of filamin A domain deletion mutants into the iPSCs to clarify the roles of interacting molecules. They found that the overexpression of wild-type filamin A restored normal thrombopoiesis, but filamin A that had domains for binding with αIIbβ3 or ρ-GTPase deleted did not. Finally, inhibitors of the RhoA effector ROCK1/2 reversed the mutation phenotypes of filamin A, in accordance with ROCK inhibitors contributing to the enhanced production of iPSC-derived platelets ex vivo.7 These findings suggest that the αIIbβ3-filamin A interaction suppresses RhoA activation to form actin stress fibers that hinder PPF. At the same time, because the deletion of RhoA results in macrothrombocytopenia in mice,8 an optimal level of RhoA activation may be required for proper PPF.
In platelets, vascular von Willibrand factor (vWF) binding to GPIb/V/IX or extracellular ADP and TXA2 binding to platelet receptors leads to a conformational change of αIIbβ3 (GPIIb/IIIa; CD41/CD61). This regulation is known as “inside-out signaling” and leads to αIIbβ3 binding with fibrinogen. Then “outside-in signaling” is triggered to induce cytoskeletal rearrangements for platelet spreading, granule secretion, and RhoA-dependent clot stabilization (see figure). Filamin A binds to β3 integrin to inhibit outside-in signaling by competing with talin or by other mechanisms.6 Conversely, in MKs, the study by Donada et al suggests that the binding of αIIbβ3 to fibrinogen, possibly in the bone marrow sinusoid or lung capillary vessels, and the suppression of RhoA activity by filamin A lead to coordinated PPF and platelet release. This mechanism may also underlie the phenotype of mutations in ITGA2B and ITGB3 to cause macrothrombocytopenia.2,9
Overall, the article by Donada et al provided novel insights into filamin A–related thrombopoiesis and revealed potential drug targets for this disease. However, the clinical application of ROCK inhibitors for congenital macrothrombocytopenia is challenging because the inhibitors may also induce chromosome instability. Drugs that specifically target the filamin A–induced RhoA activation in MKs to ameliorate thrombocytopenia in filamin A–mutant patients would be a better option. High throughput drug screening using iPSC-based MKs could be useful for identifying such drugs.10 Further studies using isogenic iPSC models of various congenital macrothrombocytopenia should help clarify the complex molecular regulation of PPF through cytoskeleton rearrangement and provide guidance for novel treatment of congenital thrombocytopenia and ex vivo platelet production.
Conflict-of-interest disclosure: K.E. is a founder of Megakaryon Co. Ltd. N.S. declares no competing financial interests.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal