

In this issue of Blood, report that loss of plant homeodomain finger 6 (Phf6) increases the ability of hematopoietic stem and progenitor cells (HSPCs) to sustain long-term engraftment and can specifically synergize with the oncogenic homeobox transcription factor TLX3 to cause lymphoid neoplasms.1
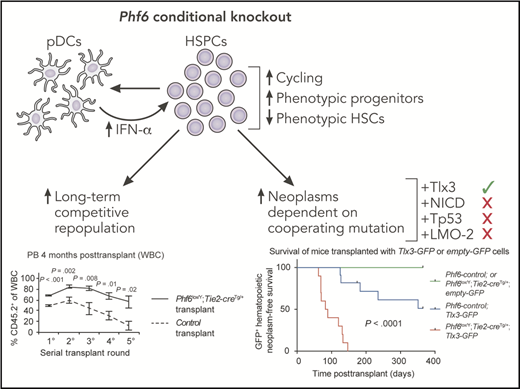
Conditional knockout of Phf6 causes increased cycling of HSPCs as well as increased phenotypically defined progenitors and decreased phenotypic hematopoietic stem cells (HSCs). These effects are partially mediated by increased production of interferon (IFN)-α by Phf6 knockout pDCs. Functionally, these alterations are associated with increased long-term competitive repopulation and increased hematologic neoplasms, the latter being dependent upon the cooperating mutation. GFP, green fluorescent protein; PB, peripheral blood; WBC, white blood cell. This figure has been adapted from Figures 4B and 7A in the article by McRae et al that begins on page 1729. Professional illustration by Patrick Lane, ScEYEnce Studios.
Conditional knockout of Phf6 causes increased cycling of HSPCs as well as increased phenotypically defined progenitors and decreased phenotypic hematopoietic stem cells (HSCs). These effects are partially mediated by increased production of interferon (IFN)-α by Phf6 knockout pDCs. Functionally, these alterations are associated with increased long-term competitive repopulation and increased hematologic neoplasms, the latter being dependent upon the cooperating mutation. GFP, green fluorescent protein; PB, peripheral blood; WBC, white blood cell. This figure has been adapted from Figures 4B and 7A in the article by McRae et al that begins on page 1729. Professional illustration by Patrick Lane, ScEYEnce Studios.
Large-scale sequencing has defined the molecular alterations in hematologic malignancies at an unprecedented resolution. With the now extensive catalog of genes that are recurrently mutated across different patients, both within and across different types of hematologic malignancies, a major challenge is to determine whether these alterations are necessary and/or sufficient for the establishment or growth of cancers in vivo. One such gene, PHF6, is one of the most frequently mutated genes in T-cell acute lymphoblastic leukemia (T-ALL)2 and is also mutated in a variety of other hematologic malignancies, including myeloid neoplasms3,4 and mixed phenotype acute leukemia.5,6 Conflicting literature on the role of PHF6 in human vs mouse malignancies7,8 has led to a major question: Is PHF6 a tumor suppressor, an oncoprotein, or a benign passenger mutation in hematologic malignancy? By using genetically engineered mouse models, a powerful tool to address this question and determine mechanism(s) of action, McRae et al show that PHF6 is a context-dependent tumor suppressor and regulates normal HSPC function (see figure).
PHF6 is a conserved, nuclear protein involved in chromatin-mediated transcriptional regulation.9 In addition to somatic mutations that are observed in hematologic malignancies, germ line mutations in PHF6 cause Börjeson-Forssman-Lehmann syndrome (BFLS), an X-linked disorder characterized by intellectual disability.9 To determine whether BFLS is a cancer predisposition syndrome, McRae et al created both germ line and conditional knockout alleles of Phf6. They observed in both cases that loss of Phf6 accelerated the onset of spontaneous hematologic neoplasms that develop in aging mice, including T-cell, B-cell, myeloerythroid, and mixed-lineage leukemias. These findings indicate that individuals with somatic and germ line mutations in PHF6 are at greater risk of hematologic malignancy.
Leukemia-associated somatic mutations, particularly those that occur early in disease development, can endow HSPCs with increased competitive fitness. McRae et al used their Phf6 conditional knockout mouse to test this concept. Although they observed little effect on the frequency of mature hematopoietic cells, they report significant alterations in the HSPC compartment, including fewer phenotypic HSCs and increased lymphoid-primed multipotent progenitors (Flt3+ hematopoietic progenitor cells [HPCs]) and common lymphoid progenitor cells. In contrast to these phenotypic alterations, serial bone marrow transplantation revealed increased long-term reconstitution of repopulating cells from Phf6 conditional knockout mice. Paradoxically, further examination of self-renewal and differentiation potential of Phf6 knockout HSCs revealed decreased self-renewal and increased differentiation. These findings may indicate that loss of Phf6 uncouples in vivo functional potential from expression of cell surface markers used to define these populations or that loss of Phf6 endows progenitor cells with long-term self-renewal ability.
To gain insight into the molecular mechanisms by which loss of Phf6 alters HSPCs, the authors performed RNA sequencing on sorted HSPC populations and uncovered signatures of increased IFN signaling. To test whether increased IFN signaling was responsible for the phenotypes observed in Phf6 conditional knockout mice, they crossed their mice with a knockout of the IFN-α receptor gene Ifnar1. Loss of Ifnar1 reversed some but not all of the phenotypes observed in Phf6 conditional knockout mice. Specifically, increase in Flt3+ HPCs and increased HSPC cycling were IFN-α dependent, whereas decrease in phenotypic HSCs and increased competitive bone marrow repopulation were independent of IFN-α signaling. Mechanistically, the authors determined that a modest increase in IFN-α levels occurred in the Phf6 conditional knockout mice, likely mediated by increased numbers of plasmacytoid dendritic cells (pDCs) that are the major producers of IFN-α in the bone marrow. These findings indicate that both HSPC-intrinsic and extrinsic mechanisms (increased production of IFN-α by pDCs) contribute to alterations in HSPCs that occur upon loss of Phf6.
To directly address conflicting results in the literature on the role of PHF6 in human vs mouse malignancies,7,8 the authors tested whether conditional knockout of Phf6 can synergize with other oncogenes or tumor suppressors to cause hematologic malignancy. They find that overexpression of Tlx3 in Phf6 conditional knockout hematopoietic cells dramatically increases penetrance and rate of leukemia development. As a caveat, these mice develop B-cell leukemia, as opposed to T-ALL, in which TLX3 mutations are typically observed in humans. This may be a consequence of the experimental design in which Tlx3 was overexpressed in HSCs rather than specifically in T cells as observed in T-ALL. In contrast to synergy with Tlx3, loss of Phf6 did not synergize with overexpression of the Notch intracellular domain, mutant Tp53, or overexpression of LMO2 to accelerate development of hematologic malignancy. These findings indicate a remarkable context specificity of tumor suppression by PHF6, dependent on the cooperating mutation(s) involved.
Considering these novel findings on context-specific tumor suppressive function of PHF6, individuals with somatic or germ line PHF6 mutations (ie, patients with BFLS) may be at risk for hematologic malignancy and likely should be monitored to enable early detection. A key outstanding question is what are the complements of cooperating mutations that do, or do not, synergize with PHF6 to cause malignancy? The cohort of spontaneous malignancies created by McRae et al offer a good resource to begin mutation profiling. In addition to molecular specificity, there is also cell-context specificity that should be addressed in future studies to determine whether the same molecular aberrations in PHF6 and cooperating alleles cause distinct hematologic malignancies in different cell types (eg, HSCs vs T cells). Finally, gaining a better understanding of the non-IFN-α–mediated HSPC-intrinsic mechanisms impacted by PHF6 mutation that function to endow these cells with a competitive advantage will be important for devising therapeutic strategies to remove those cells at risk of developing into a future malignancy.
Conflict-of-interest disclosure: J.J.T. holds a patent licensed by and receives royalties from Fate Therapeutics.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal