

TO THE EDITOR:
Waldenstrom macroglobulinemia (WM) is a rare lymphoplasmacytic lymphoma associated with immunoglobulin M monoclonal gammopathy.1 The majority of patients carry the L265P mutation in MYD88,2 whereas 40% of patients carry mutations in CXCR4.3 Mutation status has an impact on response to treatment with ibrutinib, ixazomib, and everolimus, with CXCR4-mutated WM cases demonstrating resistance to these agents.4-6
Bortezomib, a proteasome inhibitor that is the mainstay of treatment of multiple myeloma, is also effective in WM7 ; however, its relation to CXCR4 mutation status in WM has not been fully explored. In this study, we describe the impact of CXCR4 mutations on progression-free survival and overall survival of WM patients under treatment with a bortezomib-based combination and attempt to frame that in the context of pan-cancer data on CXCR4 expression and bortezomib sensitivity.
Sixty-three patients with WM treated with bortezomib/rituximab in the upfront or relapsed/refractory setting as part of a phase 2 clinical trial (www.clinicaltrials.gov, #NCT00422799) were included in this study. Patients received bortezomib IV weekly at 1.6 mg/m2 on days 1, 8, and 15, every 28 days × 6 cycles, and rituximab 375 mg/m2 at days 1, 8, 15, and 22, on cycles 1 and 4. Patients with progressive disease after 2 cycles were taken off therapy, whereas those with stable or responsive disease continued on therapy for a total of 6 cycles. Response was assessed in relation to C1D1 immunoglobulin M or M-spike measurements.8,9
Genomic DNA was extracted from formalin-fixed paraffin-embedded bone marrow, cytogenetic pellets, or CD19+ cells, selected using magnetic bead isolation (Miltenyi) from marrow aspirates (QIAmp). Mutation status of MYD88 was determined using a locked nucleic acid–blocked polymerase chain reaction method and Sanger sequencing, as previously reported.10 CXCR4 was amplified using the TTCCTTGGAGCCAAATTTAAAACC-GCTGGAGTGAAAACTTGAAGACTCA(F-R) primers, and cycling conditions were as follows: 95°C for 2 minutes, 40 cycles × (95°C for 45 seconds, 58°C for 45 seconds, 72°C for 1 minute), 72°C for 7 minutes, and 4°C. The same primer combination was used for Sanger sequencing of CXCR4 amplicons.
Twenty patients were excluded from this study because of lack of material for genotyping; however, their clinical characteristics were not different from those of the patients included (data not shown). Mutation status of MYD88 was assessed in 43 patients. DNA quality was inadequate for successful genotyping in 3 of those. Thirty-eight patients were found positive for L265P (95%), and 2 patients were found to be wild-type (WT). The WT cases had 50% and 30% lymphoplasmacytic infiltration on their bone marrow biopsy reports, and thus results were deemed reliable. Out of 43 patients that were genotyped for CXCR4, 17 (39.5%) had a mutation, which is well within the range that has been reported before.3 All patients carrying CXCR4 mutations also had MYD88 L265P. Ten out of 17 patients had frameshift mutations, 1 patient had a nonsense mutation, and 6 patients had missense mutations, all of them affecting the C-terminal portion of the protein (Table 1). The significance of missense CXCR4 mutations in patients with WM has not been determined; however, we observed 2 missense mutations, G336R and S339F, that appeared in multiple patients, alone or in combination with each other, and were predicted to be damaging by PolyPhen 2.0 with scores of 1 and 0.998, respectively.11 A different missense mutation, which was reported in WM patients by another group, was predicted to be benign by the same algorithm.12 Further testing is necessary to validate the importance of missense mutations in WM. CXCR4 mutations detected in this study are described in Table 1, together with mutations that were reported previously.12-14 Out of a total of 101 mutations reported, 51 were frameshift, whereas 42 were nonsense. Although frameshift mutations are more common collectively, S338X is still the most common mutation, occurring in a total of 34 cases.
List of CXCR4 mutations reported
| Type of mutation | Nucleotide change | Amino acid change | Times reported | Described in |
|---|---|---|---|---|
| Frameshift | c.945_946insC | H315fs | 1 | 13 |
| Frameshift | c.952_953insA | T318fs | 4 | 13,14 |
| Frameshift | c.953_954delC | T318fs | 1 | 13 |
| Frameshift | c.963_964insC | R322fs | 2 | 13,14 |
| Frameshift | c.977_978insC | L326fs | 1 | 13 |
| Frameshift | c.979_985delAGATCCT | K327fs | 1 | 13 |
| Frameshift | c.982_983delAT | I328fs | 1 | 13 |
| Nonsense | c.1000 C>T | R334X | 5 | 12-14 |
| Nonsense | c.1006 G>T | G336X | 1 | 13 |
| Nonsense | c.1013 C>A | S338X | 11 | 12-14 |
| Nonsense | c.1013 C>G | S338X | 23 | 12-14, present study |
| Frameshift | c.1012_1013delT | S338fs | 2 | 12,13 |
| Frameshift | c.1012_1015delTCAT | S338fs | 1 | 13 |
| Frameshift | c.1012_1013insT | S338fs | 8 | 13,14, present study |
| Frameshift | c.1017_1018delT | S339fs | 1 | 13 |
| Frameshift | c.1022_1023insT | S341fs | 1 | 13 |
| Nonsense | c.1031_1033delCT | S344X | 1 | 13 |
| Frameshift | c.1117_1127insTGTTTCCACTG | S339fs | 1 | 12 |
| Frameshift | c.1125_1126delCT | T342fs | 3 | 12,14, present study |
| Frameshift | c.1011_1012insA | S338fs | 1 | Present study |
| Missense | c.1006 G>A | G336R | 4 | Present study |
| Missense | c.1016 C>T | S339F | 4 | Present study |
| Frameshift | c.1007delG | G336fs | 2 | Present study |
| Frameshift | c.1002_1024del | R334fs | 1 | Present study |
| Frameshift | c.1002_1003insA | G335fs | 1 | Present study |
| Frameshift | c.970delT | S324fs | 1 | Present study |
| Frameshift | c.931_932insT | T311fs | 1 | 14 |
| Frameshift | c.951_955delACCTC | T318fs | 2 | 14 |
| Frameshift | c.954_955insC | S319fs | 1 | 14 |
| Frameshift | c.958_960delTG | V320fs | 1 | 14 |
| Frameshift | c.969_970insG | S324fs | 1 | 14 |
| Frameshift | c.978_979insT | K327fs | 1 | 14 |
| Frameshift | c.984_985insT | L329fs | 1 | 14 |
| Frameshift | c.993_994insA | G332fs | 1 | 14 |
| Frameshift | c.1005_1006insT | G336fs | 1 | 14 |
| Frameshift | c.1013_1016delATCT | S338fs | 2 | 14 |
| Frameshift | c.1013_1030delATCTGTTTCCACTGAGT | S338fs | 1 | 14 |
| Frameshift | c.1015_1017delCT | S339fs | 1 | 14 |
| Frameshift | c.1020_1021delT | S341fs | 1 | 14 |
| Frameshift | c.1030_1041CTGAGTCTTC>GT | S344fs | 1 | 14 |
| Frameshift | c.1033_1035delAG | E345fs | 1 | 14 |
| Nonsense | c.997 A>T | K333X | 1 | 14 |
| Type of mutation | Nucleotide change | Amino acid change | Times reported | Described in |
|---|---|---|---|---|
| Frameshift | c.945_946insC | H315fs | 1 | 13 |
| Frameshift | c.952_953insA | T318fs | 4 | 13,14 |
| Frameshift | c.953_954delC | T318fs | 1 | 13 |
| Frameshift | c.963_964insC | R322fs | 2 | 13,14 |
| Frameshift | c.977_978insC | L326fs | 1 | 13 |
| Frameshift | c.979_985delAGATCCT | K327fs | 1 | 13 |
| Frameshift | c.982_983delAT | I328fs | 1 | 13 |
| Nonsense | c.1000 C>T | R334X | 5 | 12-14 |
| Nonsense | c.1006 G>T | G336X | 1 | 13 |
| Nonsense | c.1013 C>A | S338X | 11 | 12-14 |
| Nonsense | c.1013 C>G | S338X | 23 | 12-14, present study |
| Frameshift | c.1012_1013delT | S338fs | 2 | 12,13 |
| Frameshift | c.1012_1015delTCAT | S338fs | 1 | 13 |
| Frameshift | c.1012_1013insT | S338fs | 8 | 13,14, present study |
| Frameshift | c.1017_1018delT | S339fs | 1 | 13 |
| Frameshift | c.1022_1023insT | S341fs | 1 | 13 |
| Nonsense | c.1031_1033delCT | S344X | 1 | 13 |
| Frameshift | c.1117_1127insTGTTTCCACTG | S339fs | 1 | 12 |
| Frameshift | c.1125_1126delCT | T342fs | 3 | 12,14, present study |
| Frameshift | c.1011_1012insA | S338fs | 1 | Present study |
| Missense | c.1006 G>A | G336R | 4 | Present study |
| Missense | c.1016 C>T | S339F | 4 | Present study |
| Frameshift | c.1007delG | G336fs | 2 | Present study |
| Frameshift | c.1002_1024del | R334fs | 1 | Present study |
| Frameshift | c.1002_1003insA | G335fs | 1 | Present study |
| Frameshift | c.970delT | S324fs | 1 | Present study |
| Frameshift | c.931_932insT | T311fs | 1 | 14 |
| Frameshift | c.951_955delACCTC | T318fs | 2 | 14 |
| Frameshift | c.954_955insC | S319fs | 1 | 14 |
| Frameshift | c.958_960delTG | V320fs | 1 | 14 |
| Frameshift | c.969_970insG | S324fs | 1 | 14 |
| Frameshift | c.978_979insT | K327fs | 1 | 14 |
| Frameshift | c.984_985insT | L329fs | 1 | 14 |
| Frameshift | c.993_994insA | G332fs | 1 | 14 |
| Frameshift | c.1005_1006insT | G336fs | 1 | 14 |
| Frameshift | c.1013_1016delATCT | S338fs | 2 | 14 |
| Frameshift | c.1013_1030delATCTGTTTCCACTGAGT | S338fs | 1 | 14 |
| Frameshift | c.1015_1017delCT | S339fs | 1 | 14 |
| Frameshift | c.1020_1021delT | S341fs | 1 | 14 |
| Frameshift | c.1030_1041CTGAGTCTTC>GT | S344fs | 1 | 14 |
| Frameshift | c.1033_1035delAG | E345fs | 1 | 14 |
| Nonsense | c.997 A>T | K333X | 1 | 14 |
Next, we assessed the impact of CXCR4 mutation status on survival of WM patients. Progression-free survival was defined as the time elapsed between time of therapy initiation and tumor progression or death, whereas overall survival was defined as the time elapsed between time of therapy initiation and death or time of last follow-up. For survival analysis, the Kaplan-Meier method was used, and differences between the curves were tested by log-rank test.
In our cohort (n = 43, upfront = 14, relapsed/refractory = 29), with a median follow-up of 90.7 months (48.4-114.3 months), CXCR4 status had no effect on progression-free survival or overall survival (log-rank, P = .994 and P = .407, respectively; Figure 1A-B). This held true even when we repeated the analysis, after excluding 6 patients with missense mutations or accounting for different treatment setting. CXCR4-mutant patients did not differ from CXCR4-WT patients in terms of toxicity or clinical data, except for lactate dehydrogenase, which was significantly lower in mutant cases. Lactate dehydrogenase largely fell within the normal range in both groups, disputing the clinical significance of this difference, while it did not affect survival, and its inclusion in the Cox regression model did not change our results.
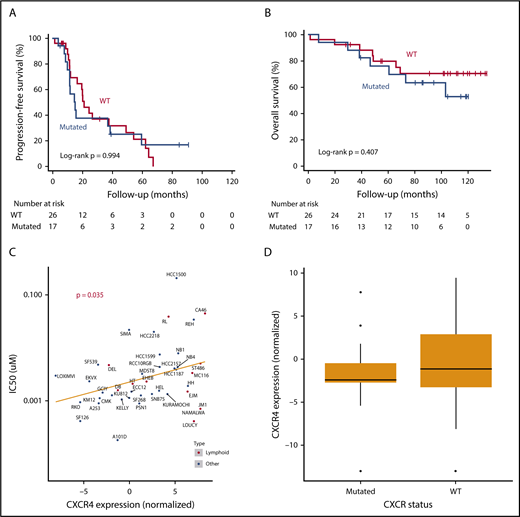
CXCR4 mutations have no effect on Waldenstrom patient survival under bortezomib-based treatment. (A) Kaplan-Meier curve of progression-free survival in CXCR4-mutated vs WT Waldenstrom macroglobulinemia patients. (B) Kaplan-Meier curve of overall survival in CXCR4-mutated vs WT Waldenstrom macroglobulinemia patients. (C) Bortezomib 50% inhibitory concentration (IC50; μM) over normalized CXCR4 expression levels in human cell lines. (D) Association of CXCR4 mutation status with levels of CXCR4 expression.
CXCR4 mutations have no effect on Waldenstrom patient survival under bortezomib-based treatment. (A) Kaplan-Meier curve of progression-free survival in CXCR4-mutated vs WT Waldenstrom macroglobulinemia patients. (B) Kaplan-Meier curve of overall survival in CXCR4-mutated vs WT Waldenstrom macroglobulinemia patients. (C) Bortezomib 50% inhibitory concentration (IC50; μM) over normalized CXCR4 expression levels in human cell lines. (D) Association of CXCR4 mutation status with levels of CXCR4 expression.
WM patients with CXCR4 mutations have historically been associated with worse response to ibrutinib and were recently shown to exhibit inferior progression-free survival, too, although a prospective trial found no effect, consistent with our bortezomib response results.4,15-17 We report for the first time that a bortezomib-based combination is impervious to the impact of CXCR4 mutations in a cohort of patients with WM. Previously, we had shown this to be true in WM cell lines, whereby genetically engineering BCWM.1 and MWCL-1 to overexpress CXCR4 had no impact on bortezomib resistance.18 Interestingly, however, that is at odds with a pan-cancer pattern of resistance to bortezomib in cell lines with higher levels of CXCR4 expression (Cancer Cell Line Encyclopedia [CCLE]19 ; Figure 1C). That being said, in the CCLE data set, there is no clear association between CXCR4 mutation status and CXCR4 expression level, as is the case in WM (Figure 1D). This indicates that other factors must drive high CXCR4 expression in the cell lines included in that data set. In fact, only 1 out of 31 mutations in CCLE affects the C-terminal portion of the protein, which is in clear contrast with what we know to be true in WM. It is thus possible that different underlying biology could explain the discrepancy between WM and other cancer types regarding the association of CXCR4 and bortezomib resistance. Our patient data indicate that survival of patients with CXCR4-mutated WM can be sustained by bortezomib-based treatment, through a mechanism that is potentially different from that of other cancers.
Different experiments have linked CXCR4 expression and bortezomib in a variety of ways in other hematological malignancies, including multiple myeloma.20-22 However, despite the complicated association in those cancer types, in WM there seems to be a consistently neutral effect of CXCR4 mutations on bortezomib resistance in both cell line and patient data. Moreover, the same holds true in patient data for another proteasome inhibitor, carfilzomib.23 Thus, our study’s findings might justify the use of bortezomib-based treatment in WM patients with CXCR4 mutations, although a prospective trial is still required for reliable conclusions. Of note, rituximab might also have played a role in these results, and so their combination should be considered. Our results also set a standard against which new CXCR4-targeting drugs, like ulocuplumab, can be compared and call for further investigation of the different mechanisms that mediate bortezomib resistance in cancer types with high CXCR4 expression.
Acknowledgments
The authors thank all the patients and their families for their contribution to this study.
This work was supported in part by the National Cancer Institute, National Institutes of Health (grant R01 CA205954), the Leukemia and Lymphoma Society, and the International Waldenström Macroglobulinemia Foundation. I.M.G. is a Scholar in Clinical Research of the Leukemia & Lymphoma Society. The authors acknowledge the generous support of the Michele & Stephen Kirsch Fund for Waldenström Macroglobulinemia (I.M.G.).
This study was approved by the Dana-Farber Cancer Institute Institutional Review Board (#06-008).
Authorship
Contribution: R.S.-P., M.C., and I.M.G. designed the research; R.S.-P., M.C., M.R., O.Z., and D.H. performed the research; R.S.-P., L.T., and C.-J.L. analyzed the data; R.S.-P. wrote the manuscript; P.H., A.S., K.R., M.B., B.R., A.P.-G., and I.M.G. cared for patients included in this study; J.J.C., S.P.T., and I.M.G. provided intellectual contributions throughout the project; and all authors reviewed and approved the manuscript.
Conflict-of-interest disclosure: I.M.G. has a consulting/advisory role with Celgene, Takeda, Bristol-Myers Squibb, Janssen Pharmaceuticals, and Amgen and has received research funding/honoraria from Celgene, Takeda, Bristol-Myers Squibb, Janssen Pharmaceuticals, and Amgen. J.J.C. received research funds from Abbvie, Beigene, Janssen, and Pharmacyclics and received honoraria from Beigene, Janssen, Merck, and Pharmacyclics. The remaining authors declare no competing financial interests.
Correspondence: Irene M. Ghobrial, Medical Oncology, Dana-Farber Cancer Institute, 450 Brookline Ave, Boston, MA 02115; e-mail: irene_ghobrial@dfci.harvard.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal