

Key Points
PRMT5 methylates and is needed for the full transcriptional repressive activity of BCL6 and is necessary for germinal center formation.
Concomitant inhibition of both BCL6 and PRMT5 exhibits synergistic killing of BCL6-expressing lymphoma cells.

Abstract
The germinal center (GC) reaction plays an important role in generating humoral immunity and is believed to give rise to most B-cell lymphomas. GC entry and exit are tightly regulated processes, controlled by the actions of transcription factors such as BCL6. Herein, we demonstrate that protein arginine methyltransferase 5 (PRMT5), a symmetric dimethyl arginine methyltransferase, is also necessary for GC formation and affinity maturation. PRMT5 contributes to GC formation and affinity maturation at least in part through its direct interaction with and methylation of BCL6 at arginine 305 (R305), a modification necessary for the full transcriptional repressive effects of BCL6. Inhibition of PRMT5 in B-cell lymphoma lines led to significant upregulation of BCL6 target genes, and the concomitant inhibition of both BCL6 and PRMT5 exhibited synergistic killing of BCL6-expressing lymphoma cells. Our studies identify PRMT5 as a novel regulator of the GC reaction and highlight the mechanistic rationale of cotargeting PRMT5 and BCL6 in lymphoma.
Introduction
The germinal center (GC) reaction plays a central role in generating high affinity B-cell responses to antigens. Functionally, GCs are divided into the dark zone, a site of B-cell hyperproliferation where the B-cell receptor repertoire is shaped by somatic hypermutation, and the light zone, where B cells with high antigen affinity are selected for survival through cognate antigenic interactions.1,2 Initiation and normal GC progression relies on coordinated changes in gene expression profiles mediated by transcription factors such as BCL6, the “master regulator” of the GC reaction.3-5 Deletion of BCL6 leads to loss of GCs, while constitutive expression of BCL6 induced through chromosomal translocations, amplifications, and/or mutations of BCL6 regulatory elements have been demonstrated in lymphomas, highlighting BCL6’s importance in driving these processes.6-11 Deregulated expression of BCL6 in mice recapitulates human diffuse large B-cell lymphoma (DLBCL) pathogenesis.12 However, how BCL6 regulates GC dynamics and lymphoid transformation remains incompletely understood, especially in regards to its functional regulation through posttranslational modifications.
BCL6 is a transcriptional repressor whose repressive activity is mediated via its N terminal BTB/POZ and middle RD2 domains.13-15 BCL6 BTB/POZ mutant mice exhibit decreased GC B-cell survival and proliferation, and although they have GCs, they are fewer and smaller than those observed in wild-type mice.16 In contrast, disruption of the RD2 domain of BCL6 leads to loss of GC formation via inhibiting B-cell migration to lymphoid follicles,17 illustrating the complementary roles these domains play in orchestrating the GC reaction. Repression by the BTB/POZ domain in BCL6 is mediated by its recruitment of the SMRT, NCOR, and BCOR corepressors,13,14,18 while repression by the RD2 domain is mediated by its recruitment of histone deacetylase–containing NuRD complexes17,19 ; nonetheless, much remains to be understood about the regulation of BCL6 activity.
Protein arginine methyltransferase 5 (PRMT5) is a member of the PRMT family of enzymes. PRMT5 forms a complex with the MEP50 protein that is required for its methyltransferase activity, allowing it to function as a transcriptional repressor. PRMT5 is the major symmetric arginine dimethyltransferase in cells, which methylates histones H3R8 and H4R320,21 as well as a variety of nonhistone protein substrates.22,23 PRMT5 is found in chromatin remodeling complexes, including the hSWI/SNF and NuRD complex.24,25 Previous studies have demonstrated that PRMT5 is expressed in GC B cells and malignant lymphomas and that inhibition of PRMT5 activity suppresses lymphoma cell proliferation and induces apoptosis.26-28
Given that PRMT5 plays an important role in maintaining the growth of GC-derived lymphomas and that it associates with the NuRD complex, which contributes to the repressive activity of BCL6,16,19 we hypothesized that PRMT5 may play an important role in GC formation and lymphomagenesis, at least in part by regulating BCL6 function. Using a variety of approaches, including genetically engineered, conditional knockout mice and biochemical studies, we show that PRMT5 is required for GC formation, and it directly binds BCL6 and regulates BCL6 repressive function through arginine methylation. We also demonstrate synergistic killing of lymphomas by concomitant inhibition of BCL6 and PRMT5. This may illuminate a mechanism that is critically important for GC formation and the proliferation of GC-derived B-cell lymphomas.
Materials and methods
Cells and reagents
The non-Hodgkin lymphoma cell lines Raji, SUDHL8, U2932, SUDHL4, and SUDHL6 were cultured in RPMI 1640 medium (Fisher Scientific, Santa Clara, CA). OCI-LY1 and OCI-LY7 cells were cultured in IMDM (Fisher Scientific) with 20% human AB serum (Valley Biomedical, Winchester, VA). 293T cells were maintained in Dulbecco’s modified Eagle medium (Gibco, Grand Island, NY). All types of cell culture medium were supplemented with 10% fetal bovine serum (unless stated otherwise), 100 µg/mL penicillin, 100 U/mL streptomycin, and 2 mM l-glutamine (Fisher Scientific).
The GAL4 expression vector,29 pGBX1 BCL6, pGBX1 BTB, pGBX1 RD2,17 and pcDNA3 hemagglutinin (HA) PRMT530 plasmids were previously described. The synthetic Bcl6 reporter, B6BS-TK-Luc, which contains 4 copies of consensus Bcl6 DNA-binding sites,31 was a gift of Hilda Ye. pcDNA3.1 HA PRMT5(1-320), pCMV-Sport 6 MEP50, pcDNA3.1 V5 BCL6, pcDNA3.1 V5 BTB, pcDNA3.1 V5 RD2, pcDNA3.1 V5 ZF, and pcDNA3.1 V5 PRMT5 G367A/R368A were generated in our laboratory using standard molecular cloning techniques and confirmed by sequencing.
Recombinant glutathione S-transferase (GST) PRMT5 protein was from Novus Biologicals (Littleton, CO), recombinant BCL6 was from Origene (Rockville, MD), recombinant GST-MEP50 was from Abnova (Walnut, CA), and recombinant human histone H2A and S-adenosyl-methionine (SAM) were from New England Biolabs (Ipswich, MA). His-tagged RD2 protein was purified by using the Takara HisTALON Superflow Cartridge Purification Kit (Mountain View, CA). BCL6 (C-19; sc-368), BCL6 (N-3), MEP50 (C-2; sc-376549), GFP (B-2; sc-9996), and GST (1E5; sc-53909) antibodies were from Santa Cruz Biotechnology (Dallas, TX). PRMT5 (07-405), SYM10 (07-412), β-actin (AC-15; A5441), HA (HA-7; H3663) and the ASYM24 antibody were from MilliporeSigma (St. Louis, MO). Monomethyl arginine (D5A12) and symmetric dimethyl arginine (SDMA) (SDMA motif [sdme-RG] MultiMab rabbit monoclonal antibody) antibodies were from Cell Signaling Technology (Danvers, MA). The V5 antibody (R960-25) and PRMT5 and MEP50 small interfering RNAs (siRNAs) were from Thermo Fisher Scientific (Waltham, MA).
Murine models
The Research Animal Resource Center of the Weill Cornell College of Medicine approved all mouse procedures. The generation of conditional Prmt5 knockout (Prmt5fl/fl) mice has been previously reported.34 GC-specific conditional knockout of Prmt5 was generated by crossing Prmt5fl/fl mice with the Cγ1-Cre strain (The Jackson Laboratory, 010611) to produce heterozygous mice, which were then crossed to yield Cγ1-Cre+/−Prmt5fl/fl. Cγ1-Cre−/−Prmt5fl/fl littermates were used as a control group.
Statistical analysis
A 2-tailed Student t test and a Welch unequal variances t test were used, and a P < .05 was considered statistically significant.
Additional methods are described in supplemental Materials and methods (available on the Blood Web site). The data from the RNA-seq experiments are available at the Gene Expression Omnibus database (http://www.ncbi.nlm.nih.gov/geo/) with accession number GSE116963.
Results
PRMT5 is required for GC formation and is necessary for immunoglobulin affinity maturation
Given that PRMT5 is found in protein complexes that regulate the repressive activity of BCL6, a master regulator of the GC reaction, we hypothesized that it also plays an important function in GC B cells. We first sought to determine if PRMT5 was required for GC formation. Constitutive Prmt5 deletion is embryonic lethal.35 Therefore, to determine if Prmt5 is required for the GC reaction, we crossed Prmt5-floxed (Prmt5fl/fl) mice34 with Cγ1-Cre mice to generate specific conditional deletion of Prmt5 in GC B cells. To induce GC formation, we immunized these mice with a T-cell–dependent antigen, sheep red blood cells (SRBCs), and sacrificed these animals 10 days later when GC formation is at its peak. Cγ1-Cre+/−Prmt5fl/fl mice displayed significantly smaller GCs in the spleen (P = .0026), as assayed by immunohistochemical staining for peanut agglutinin (PNA), a GC B cell marker, as well as reduced proliferation, measured by Ki-67 staining, compared with Cγ1-Cre−/−Prmt5fl/fl mice (Figure 1A-B). Residual GC B cells are likely due to incomplete Cre-mediated excision, as several GC clusters stained positive for Prmt5 (supplemental Figure 1). Concordantly we also observed a significant decrease in the frequency of splenic GC B-cells (GL7+/FAS+/B220+ and CD38dim/FAS+/B220+) of Cγ1-Cre+/−Prmt5fl/fl mice by flow cytometry compared with Cγ1-Cre−/−Prmt5fl/fl (Figure 1C).
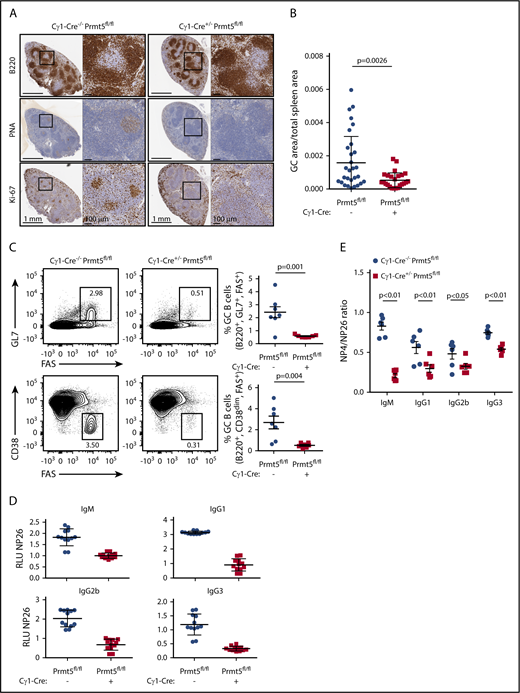
PRMT5 is required for GC formation and affinity maturation. (A) Immunohistochemistry of paraffin-embedded splenic tissue from Cγ1-Cre−/−Prmt5fl/fl and Cγ1-Cre+/−Prmt5fl/fl mice (n = 7 per group) immunized with SRBCs for 10 days. (B) Quantification of PNA+ clusters from panel A. (C) Representative flow cytometry plot showing percentage of GC B cells (GL7+ FAS+ or CD38dim FAS+) gated on live B220+ splenocytes in Cγ1-Cre−/−Prmt5fl/fl and Cγ1-Cre+/−Prmt5fl/fl mice (n = 7 per group) immunized with SRBC as described in panel A. (D) Titers of low-affinity NP-specific immunoglobulin were measured using NP26-BSA in the serum of Cγ1-Cre−/−Prmt5fl/fl and Cγ1-Cre+/−Prmt5fl/fl mice (n = 6 per group) immunized with NP-CGG for 8 days. (E) Ratio of high- to low-affinity NP-specific immunoglobulin detected with NP4-BSA and NP26-BSA, respectively, in NP-CGG immunized Cγ1-Cre−/−Prmt5fl/fl and Cγ1-Cre+/−Prmt5fl/fl mice described in panel D.
PRMT5 is required for GC formation and affinity maturation. (A) Immunohistochemistry of paraffin-embedded splenic tissue from Cγ1-Cre−/−Prmt5fl/fl and Cγ1-Cre+/−Prmt5fl/fl mice (n = 7 per group) immunized with SRBCs for 10 days. (B) Quantification of PNA+ clusters from panel A. (C) Representative flow cytometry plot showing percentage of GC B cells (GL7+ FAS+ or CD38dim FAS+) gated on live B220+ splenocytes in Cγ1-Cre−/−Prmt5fl/fl and Cγ1-Cre+/−Prmt5fl/fl mice (n = 7 per group) immunized with SRBC as described in panel A. (D) Titers of low-affinity NP-specific immunoglobulin were measured using NP26-BSA in the serum of Cγ1-Cre−/−Prmt5fl/fl and Cγ1-Cre+/−Prmt5fl/fl mice (n = 6 per group) immunized with NP-CGG for 8 days. (E) Ratio of high- to low-affinity NP-specific immunoglobulin detected with NP4-BSA and NP26-BSA, respectively, in NP-CGG immunized Cγ1-Cre−/−Prmt5fl/fl and Cγ1-Cre+/−Prmt5fl/fl mice described in panel D.
We next asked whether the impaired GC reaction observed in Cγ1-Cre+/−Prmt5fl/fl mice is able to successfully generate class-switched high-affinity immunoglobulins. To test this, we immunized Cγ1-Cre+/−Prmt5fl/fl and Cγ1-Cre−/−Prmt5fl/fl mice with 4-hydroxy-3-nitrophenylacetyl-chicken γ-globulin (NP-CGG) to induce cognate high-affinity antibodies. Compared with Cγ1-Cre−/−Prmt5fl/fl mice, Cγ1-Cre+/−Prmt5fl/fl mice developed significantly fewer low-affinity (NP26-reactive) immunoglobulin M (IgM) and class-switched IgG1, IgG2b, and IgG3 isotype B cells, consistent with the observed GC hypoplasia (Figure 1D). Even more strikingly, immunized Cγ1-Cre+/−Prmt5fl/fl mice generated much lower titers of high-affinity (NP4-reactive) IgM, IgG1, IgG2b, and IgG3, which was evident by a decreased ratio of NP4-BSA to NP26-BSA titers (Figure 1E). Taken together, these observations demonstrate that Prmt5 is necessary for GC formation and immunoglobulin affinity maturation.
PRMT5 directly interacts with BCL6 via the BCL6 RD2 and ZNF domains
Constitutive or GC-specific loss of Bcl6 leads to decreased GC number and affinity maturation defects,6,7,11 a phenotype similar to what we observed with deletion of Prmt5. We therefore explored whether PRMT5 interacts with and regulates BCL6 function. To test this, we transfected BCL6 into 293T cells and performed immunoprecipitation (IP) using an anti-BCL6 antibody followed by western blot for endogenous PRMT5, which showed that BCL6 interacts with PRMT5 (Figure 2A). The interaction was confirmed by a reciprocal IP for PRMT5 and western blot for BCL6. To test if this interaction is also observed in B cells expressing endogenous BCL6, we repeated the IP experiments in the DLBCL cell lines U2932 and SUDHL8 and again demonstrated the PRMT5-BCL6 interaction (Figure 2B). This interaction was also observed following Benzonase pretreatment, suggesting that the PRMT5-BCL6 interaction does not require the presence of nucleic acids (supplemental Figure 2). Critically, the endogenous BCL6-PRMT5 interaction was also observed in primary human GC B cells (Figure 2C), highlighting the potential physiological importance of this protein interaction.
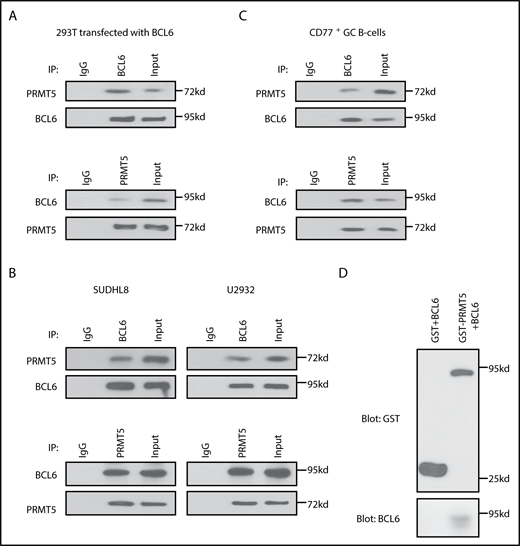
The arginine methyltransferase PRMT5 directly interacts with BCL6. (A) Co-IP experiments for PRMT5 and BCL6 in 293T cells transfected with BCL6. (B) Co-IP experiments for PRMT5 and BCL6 in SUDHL8 and U2932 DLBCL cell lines. (C) PRMT5 and BCL6 interact in normal CD77+ GC B cells, enriched as described in supplemental Materials and methods. (D) GST pull-down assay of recombinant GST-PRMT5 and BCL6-MYC/DDK proteins. Purified GST-PRMT5 or GST proteins were incubated with BCL6-MYC/DDK protein for 12 hours. The coprecipitated BCL6 and PRMT5 proteins were detected by western blot with anti-GST and anti-BCL6 antibodies.
The arginine methyltransferase PRMT5 directly interacts with BCL6. (A) Co-IP experiments for PRMT5 and BCL6 in 293T cells transfected with BCL6. (B) Co-IP experiments for PRMT5 and BCL6 in SUDHL8 and U2932 DLBCL cell lines. (C) PRMT5 and BCL6 interact in normal CD77+ GC B cells, enriched as described in supplemental Materials and methods. (D) GST pull-down assay of recombinant GST-PRMT5 and BCL6-MYC/DDK proteins. Purified GST-PRMT5 or GST proteins were incubated with BCL6-MYC/DDK protein for 12 hours. The coprecipitated BCL6 and PRMT5 proteins were detected by western blot with anti-GST and anti-BCL6 antibodies.
We next asked whether the interaction between PRMT5 and BCL6 was direct. GST pull-down assays of recombinant GST-PRMT5 and recombinant BCL6 demonstrated direct binding between these proteins (Figure 2D). We next asked what domains of BCL6 mediate its interaction with PRMT5. To this end, we transfected 293T cells with plasmids encoding V5-tagged full-length BCL6 as well as individual BTB/POZ, RD2, and ZNF domains (Figure 3A). We performed IPs with PRMT5 and demonstrated an interaction of PRMT5 with the BCL6 RD2 and ZNF domains (Figure 3B). This interaction was confirmed by reciprocal IP for the V5 tag and blotting for PRMT5. We confirmed a direct interaction between these domains by performing GST pull-down assays of recombinant GST-PRMT5 and a His-tagged RD2 domain of BCL6 (Figure 3C). A similar V5 pull-down assay of recombinant GST-PRMT5 and a V5-tagged ZNF domain of BCL6 also demonstrated direct interaction between these proteins (Figure 3D).
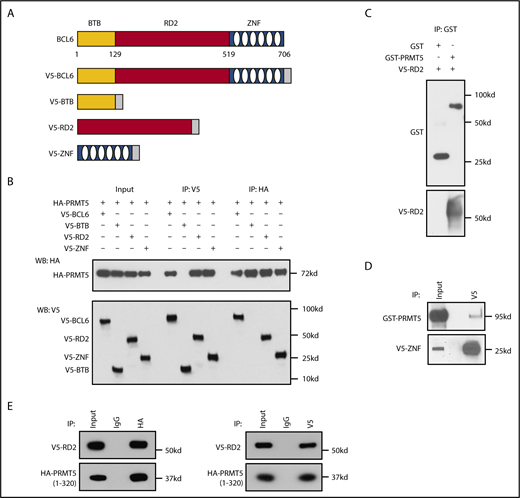
PRMT5 interacts with the RD2 and ZNF domains of BCL6 via its N terminus. (A) Schematic of plasmids encoding either the V5-tagged full-length BCL6 or domains of BCL6 used in panel B. (B) Co-IP experiments of PRMT5 with V5-tagged full-length or BTB/POZ, RD2 and ZNF domains of BCL6. (C) GST pull-down assay of recombinant GST-PRMT5 with BCL6 RD2-V5/His proteins. (D) V5 pull-down assay of recombinant GST-PRMT5 with BCL6 ZNF-V5 proteins. (E) RD2 domain of BCL6 interacts with the catalytically active N terminus of PRMT5 (residues 1-320).
PRMT5 interacts with the RD2 and ZNF domains of BCL6 via its N terminus. (A) Schematic of plasmids encoding either the V5-tagged full-length BCL6 or domains of BCL6 used in panel B. (B) Co-IP experiments of PRMT5 with V5-tagged full-length or BTB/POZ, RD2 and ZNF domains of BCL6. (C) GST pull-down assay of recombinant GST-PRMT5 with BCL6 RD2-V5/His proteins. (D) V5 pull-down assay of recombinant GST-PRMT5 with BCL6 ZNF-V5 proteins. (E) RD2 domain of BCL6 interacts with the catalytically active N terminus of PRMT5 (residues 1-320).
We next asked whether the N-terminal region of PRMT5, which contains its catalytic domain, mediates the interaction with the BCL6 RD2 domain. To test this, we generated an HA-tagged N terminus construct of PRMT5 (residues 1-320) and repeated the IP experiments. We observed that PRMT5 (1-320) interacted with the BCL6 RD2 domain (Figure 3E). Collectively, these data reveal the existence of a direct BCL6-PRMT5 protein-protein interaction.
PRMT5 facilitates BCL6-mediated transcriptional repression
BCL6 and PRMT5 have both been demonstrated to function as transcriptional repressors, and given their direct interaction, we asked whether PRMT5 was necessary for BCL6 to exert its transcriptional repressive activity. To test this, we used 2 different transcriptional reporter systems to monitor BCL6 repressive activity in 293T cells: (1) a GAL4-dependent luciferase reporter plasmid (Figure 4A-B) whose transcriptional activity was assessed following transfection of plasmids encoding the GAL4 DNA-binding domain (GAL4DBD) fused to BCL6 (GAL4DBD-BCL6), either alone or in combination with PRMT5 and MEP50; and (2) a 4xBCL6-binding site–driven luciferase reporter plasmid (Figure 4C) whose transcriptional activity was assessed following transfection of plasmids encoding BCL6, either alone or in combination with PRMT5 and MEP50. In both systems, BCL6 repressed the reporter constructs as expected but repressed even stronger upon cotransfection with PRMT5 and MEP50 (P < .001 for both reporters) (Figure 4B-C). In addition, while knockdown of PRMT5 or MEP50 with 2 distinct sets of siRNA for each protein decreased GAL4DBD-BCL6’s repressive activity, their combined knockdown impaired BCL6’s activity to a greater extent (Figure 4D; supplemental Figure 3; each representing distinct set of siRNAs). Concordantly, PRMT5 inhibitors, GSK591 and methylthioadenosine, also decreased BCL6’s repressive activity (Figure 4E; supplemental Figure 4). Furthermore, in contrast to the wild-type PRMT5 protein, which enhances the repressive activity of BCL6, transfection of a catalytically inactive PRMT5 mutant (G367A/R368A: muPRMT5) failed to enhance BCL6 repressive activity (Figure 4F). Together, these observations suggest that catalytically active PRMT5/MEP50 contributes to BCL6’s transcriptional repressive activity.
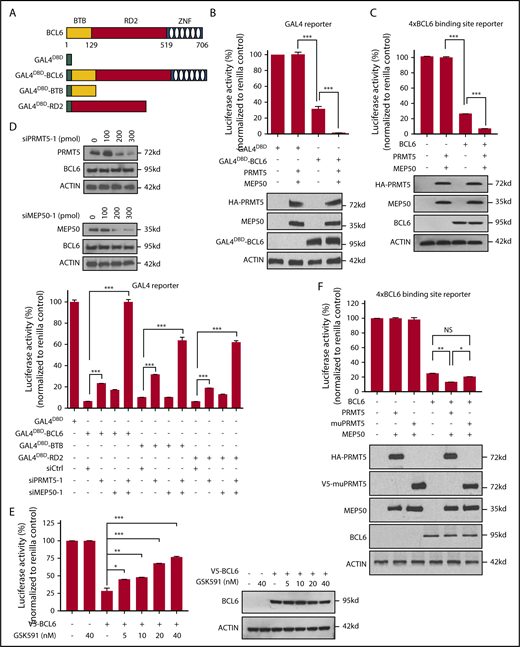
PRMT5 and MEP50 mediate the repressive activity of the BCL6 RD2 domain. (A) Schematic of plasmids encoding the GAL4 DNA-binding domain (GAL4DBD) fused to either full-length BCL6 or domains of BCL6 used in panels B and D. (B) GAL4 luciferase reporter assays in 293T cells transfected with plasmids encoding GAL4DBD-BCL6 and PRMT5/MEP50. Western blots demonstrate representative expression of GAL4DBD-BCL6, PRMT5, and MEP50 following expression of corresponding expression vectors. All the experiments were repeated 3 times in triplicate. ***P < .001. (C) 4xBCL6 binding site luciferase reporter assays in 293T cells transfected with plasmids encoding BCL6 and PRMT5/MEP50. Western blots demonstrate representative expression of BCL6, PRMT5, and MEP50 following expression of corresponding expression vectors. All the experiments were repeated 3 times in triplicate. ***P < .001. (D) GAL4 luciferase reporter assays in 293T cells transfected with full-length GAL4DBD-BCL6, GAL4DBD-BTB/POZ, or GAL4DBD-RD2 domains of BCL6 in the presence or absence of cotransfected PRMT5/MEP50 siRNAs from GE Dharmacon (Lafayette, CO). Western blots demonstrate representative expression of PRMT5 and BCL6 or MEP50 and BCL6 following transfection of increasing concentrations of PRMT5 siRNA-1 and MEP50 siRNA-1, respectively. All the experiments were repeated 3 times in triplicate. ***P < .001. Independent siRNA to PRMT5 and MEP50 shown in supplemental Figure 3. (E) 4xBCL6 binding site luciferase reporter assays in 293T cells transfected with plasmids encoding BCL6 alone or in the presence of GSK591 (*P < .05; **P < .01; ***P < .001). (F) 4xBCL6 binding site luciferase reporter assays in 293T cells transfected with plasmids encoding BCL6, PRMT5, or catalytically inactive PRMT5 mutant (G367A/R368A, muPRMT5). Western blots demonstrate representative expression of BCL6, PRMT5, and muPRMT5 (G367A/R368A) following expression of corresponding expression vectors. All the experiments were repeated 3 times in triplicate. *P < .05; **P < .01.
PRMT5 and MEP50 mediate the repressive activity of the BCL6 RD2 domain. (A) Schematic of plasmids encoding the GAL4 DNA-binding domain (GAL4DBD) fused to either full-length BCL6 or domains of BCL6 used in panels B and D. (B) GAL4 luciferase reporter assays in 293T cells transfected with plasmids encoding GAL4DBD-BCL6 and PRMT5/MEP50. Western blots demonstrate representative expression of GAL4DBD-BCL6, PRMT5, and MEP50 following expression of corresponding expression vectors. All the experiments were repeated 3 times in triplicate. ***P < .001. (C) 4xBCL6 binding site luciferase reporter assays in 293T cells transfected with plasmids encoding BCL6 and PRMT5/MEP50. Western blots demonstrate representative expression of BCL6, PRMT5, and MEP50 following expression of corresponding expression vectors. All the experiments were repeated 3 times in triplicate. ***P < .001. (D) GAL4 luciferase reporter assays in 293T cells transfected with full-length GAL4DBD-BCL6, GAL4DBD-BTB/POZ, or GAL4DBD-RD2 domains of BCL6 in the presence or absence of cotransfected PRMT5/MEP50 siRNAs from GE Dharmacon (Lafayette, CO). Western blots demonstrate representative expression of PRMT5 and BCL6 or MEP50 and BCL6 following transfection of increasing concentrations of PRMT5 siRNA-1 and MEP50 siRNA-1, respectively. All the experiments were repeated 3 times in triplicate. ***P < .001. Independent siRNA to PRMT5 and MEP50 shown in supplemental Figure 3. (E) 4xBCL6 binding site luciferase reporter assays in 293T cells transfected with plasmids encoding BCL6 alone or in the presence of GSK591 (*P < .05; **P < .01; ***P < .001). (F) 4xBCL6 binding site luciferase reporter assays in 293T cells transfected with plasmids encoding BCL6, PRMT5, or catalytically inactive PRMT5 mutant (G367A/R368A, muPRMT5). Western blots demonstrate representative expression of BCL6, PRMT5, and muPRMT5 (G367A/R368A) following expression of corresponding expression vectors. All the experiments were repeated 3 times in triplicate. *P < .05; **P < .01.
We next asked what repressor domain of BCL6 was dependent on PRMT5. To test this, we used expression constructs containing the GAL4DBD-BCL6 BTB/POZ, GAL4DBD-BCL6 RD2 and GAL4DBD-BCL6 ZNF domains (Figure 4A) and repeated the GAL4 luciferase reporter assays (Figure 4D; supplemental Figure 3; data not shown). Consistent with previous reports, we observed strong repression of luciferase activity by the BCL6 BTB and RD2 domains, but not the ZNF domain (Figure 4D; supplemental Figure 3; data not shown). When the BCL6 BTB and RD2 domains were cotransfected with siRNAs to PRMT5 and MEP50, we observed impaired repression for both domains (P < .001 for all comparisons). The observed antagonism of the BTB domain mediated repression induced by cotransfection of PRMT5 and MEP50 siRNAs was likely because PRMT5 is known to be found in complexes containing BCL6 BTB domain-interacting proteins (eg, NCOR and SMRT), since this domain did not interact directly with PRMT5.36 Overall, the PRMT5/MEP50 complex enhances the repressor activity of both BCL6 repressor domains, likely through direct and indirect mechanisms.
PRMT5 directly methylates BCL6 at R305 to regulate its transcriptional repressor activity
PRMT5 can affect transcriptional repression by its SDMA residues within a variety of histone and nonhistone proteins, which prompted us to determine whether BCL6 is symmetrically dimethylated by PRMT5. To test this, we performed IP using the SYM10 antibody and observed the presence of BCL6 in 2 independent lymphoma cell lines, which suggested that BCL6 might be symmetrically dimethylated (Figure 5A). PRMT5 knockdown using siRNA markedly decreased BCL6 symmetric arginine dimethylation without affecting overall BCL6 protein expression levels (Figure 5B). To determine whether BCL6 dimethylation is directly mediated by PRMT5, we performed two distinct in vitro methyltransferase assays with BCL6 and PRMT5/MEP50. Following in vitro incubation of recombinant BCL6 and PRMT5/MEP50 with SAM, western blot with the SYM10 antibody detected BCL6, as confirmed by BCL6 and SYM10 co-IP, suggesting that BCL6 was directly posttranslationally dimethylated by PRMT5 (Figure 5C). Furthermore, in vitro methyltransferase assays using radiolabeled 3H SAM also confirmed BCL6 arginine methylation by PRMT5 (Figure 5D). In both assays, PRMT5 also dimethylated arginine residues in histone 2A (used as a positive control).

PRMT5 directly dimethylates arginines of BCL6 in lymphoma. (A) IP for BCL6 and symmetric arginine dimethylation (SYM10) in lymphoma cell lines OCI-LY1 and Raji reveals symmetric arginine dimethylation of BCL6. (B) Knockdown of PRMT5 with specific siRNA decreases BCL6 symmetric arginine methylation. Raji cells were transfected with PRMT5 or control siRNAs followed by IP with symmetric arginine dimethylation (SYM10) antibody and immunoblotting with BCL6 antibody. Also shown are western blots with indicated antibodies from the same cells. (C) In vitro methyltransferase assay with recombinant PRMT5, MEP50, and BCL6 or H2A proteins. The proteins were blotted with the indicated antibodies. In addition, the reaction mixture was immunoprecipitated with BCL6 antibody and blotted with antibodies for symmetric arginine dimethylation (SYM10) and BCL6. (D) In vitro thymidine incorporation methyltransferase assay with recombinant PRMT5 and BCL6 or H2A proteins.
PRMT5 directly dimethylates arginines of BCL6 in lymphoma. (A) IP for BCL6 and symmetric arginine dimethylation (SYM10) in lymphoma cell lines OCI-LY1 and Raji reveals symmetric arginine dimethylation of BCL6. (B) Knockdown of PRMT5 with specific siRNA decreases BCL6 symmetric arginine methylation. Raji cells were transfected with PRMT5 or control siRNAs followed by IP with symmetric arginine dimethylation (SYM10) antibody and immunoblotting with BCL6 antibody. Also shown are western blots with indicated antibodies from the same cells. (C) In vitro methyltransferase assay with recombinant PRMT5, MEP50, and BCL6 or H2A proteins. The proteins were blotted with the indicated antibodies. In addition, the reaction mixture was immunoprecipitated with BCL6 antibody and blotted with antibodies for symmetric arginine dimethylation (SYM10) and BCL6. (D) In vitro thymidine incorporation methyltransferase assay with recombinant PRMT5 and BCL6 or H2A proteins.
We next asked which arginine residues in BCL6 are dimethylated by PRMT5. To this end, we performed mass spectroscopy analysis of BCL6 following an in vitro methyltransferase assay with BCL6 and PRMT5. Based on the number of isolated peptides, PRMT5 dimethylated BCL6 mainly at R305, located in the RD2 domain of BCL6 (Figure 6A). A few additional peptides with potential arginine dimethylation at R170 and R217 were identified. We did not observe arginine monomethylation or asymmetric dimethylation (supplemental Figure 5). To determine whether arginine dimethylation of R305, the most frequently observed dimethylated arginine of BCL6, was important for BCL6 transcriptional repressor activity, we generated a BCL6 G914A mutant, which encodes a BCL6 R305K protein that is stable and interacts with known BCL6 partners (eg, BCOR; supplemental Figure 6). We repeated the BCL6 luciferase reporter assays with the mutant BCL6 R305K protein. Compared with wild-type BCL6, BCL6 R305K did not inhibit luciferase expression (P < .001) (Figure 6B), despite the fact that it was dimethylated, likely at R170 and R217 (Figure 6C).
![Figure 6. PRMT5 dimethylates BCL6 at R305 to mediate the repressive activity of the BCL6 RD2 domain. (A) Mass spectroscopy of BCL6 protein following in vitro methyltransferase assay by PRMT5 identifies BCL6 methylation at R305. Representative tandem mass spectrometry spectrum of methylated R305 of BCL6 (P11482). Band corresponding to BCL6 was excised from the gel and subjected to trypsin digestion. Tryptic peptides were resolved on a reverse phase column, and high-energy collision dissociation spectra were obtained using Orbitrap Fusion Tribrid mass spectrometer. Data were searched against human protein database using Proteome Discoverer (v 1.4, ThermoScientific) by considering methylation on arginine as a potential modification. Results were filtered at 1% false discovery rate. A tandem mass spectrometry spectrum corresponding to 302EEErPSSEDEIALHFEPPNAPLNR325 of BCL6 (precursor [M+H]+4 = 698.3398 m/z. DPPM = 1.6, inset) is shown. Observed b- and y-ions are indicated. The lowercase “r” is the methylated arginine. (B) 4xBCL6 binding site luciferase reporter assays in 293T cells transfected with wild-type and R305K mutant BCL6 or control pcDNA3.1 vector in the presence or absence of cotransfected siRNAs to PRMT5. ***P < .001. Western blots in each experimental condition with the indicated antibodies. (C) Arginine dimethylation of wild-type or R305K BCL6 transfected into Raji cells. (D) Co-IP experiments of endogenous PRMT5 with wild-type or R305K BCL6 transfected into Raji and 293T cells. WT, wild-type.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/19/10.1182_blood-2018-02-831438/4/m_blood831438f6.png?Expires=1769152252&Signature=NCjAHq1NviP6xBp1V0Iz0yLpAcrEUVSKM8l2tePPCR7RNNN6~e1E1x8azQFsh5QNwUCAN5SpOunQEgHR2r0C9flxCC5WOda9NDqTseicYCruvVK~U-pITNgotVYs-E0WyD-3E7pbfev1DbfwercOhbekPo4EnmL1~oweSTu-UNrGyfMsOT0EbAU189hKa1LAmBPtYWv4wqijSUX72lVqO3j1d7Ty-8CbR6YwNFmDxb3Bba4lKiveihbgrFFPr9IcnIZFv111jfMKxOuzvP632-UF5PNispSXadUXp6YWCh6Fy~gYqUKgKVtagM~en8CCHvTAkWv1KjCCK5kN72NVeg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
PRMT5 dimethylates BCL6 at R305 to mediate the repressive activity of the BCL6 RD2 domain. (A) Mass spectroscopy of BCL6 protein following in vitro methyltransferase assay by PRMT5 identifies BCL6 methylation at R305. Representative tandem mass spectrometry spectrum of methylated R305 of BCL6 (P11482). Band corresponding to BCL6 was excised from the gel and subjected to trypsin digestion. Tryptic peptides were resolved on a reverse phase column, and high-energy collision dissociation spectra were obtained using Orbitrap Fusion Tribrid mass spectrometer. Data were searched against human protein database using Proteome Discoverer (v 1.4, ThermoScientific) by considering methylation on arginine as a potential modification. Results were filtered at 1% false discovery rate. A tandem mass spectrometry spectrum corresponding to 302EEErPSSEDEIALHFEPPNAPLNR325 of BCL6 (precursor [M+H]+4 = 698.3398 m/z. DPPM = 1.6, inset) is shown. Observed b- and y-ions are indicated. The lowercase “r” is the methylated arginine. (B) 4xBCL6 binding site luciferase reporter assays in 293T cells transfected with wild-type and R305K mutant BCL6 or control pcDNA3.1 vector in the presence or absence of cotransfected siRNAs to PRMT5. ***P < .001. Western blots in each experimental condition with the indicated antibodies. (C) Arginine dimethylation of wild-type or R305K BCL6 transfected into Raji cells. (D) Co-IP experiments of endogenous PRMT5 with wild-type or R305K BCL6 transfected into Raji and 293T cells. WT, wild-type.
PRMT5 dimethylates BCL6 at R305 to mediate the repressive activity of the BCL6 RD2 domain. (A) Mass spectroscopy of BCL6 protein following in vitro methyltransferase assay by PRMT5 identifies BCL6 methylation at R305. Representative tandem mass spectrometry spectrum of methylated R305 of BCL6 (P11482). Band corresponding to BCL6 was excised from the gel and subjected to trypsin digestion. Tryptic peptides were resolved on a reverse phase column, and high-energy collision dissociation spectra were obtained using Orbitrap Fusion Tribrid mass spectrometer. Data were searched against human protein database using Proteome Discoverer (v 1.4, ThermoScientific) by considering methylation on arginine as a potential modification. Results were filtered at 1% false discovery rate. A tandem mass spectrometry spectrum corresponding to 302EEErPSSEDEIALHFEPPNAPLNR325 of BCL6 (precursor [M+H]+4 = 698.3398 m/z. DPPM = 1.6, inset) is shown. Observed b- and y-ions are indicated. The lowercase “r” is the methylated arginine. (B) 4xBCL6 binding site luciferase reporter assays in 293T cells transfected with wild-type and R305K mutant BCL6 or control pcDNA3.1 vector in the presence or absence of cotransfected siRNAs to PRMT5. ***P < .001. Western blots in each experimental condition with the indicated antibodies. (C) Arginine dimethylation of wild-type or R305K BCL6 transfected into Raji cells. (D) Co-IP experiments of endogenous PRMT5 with wild-type or R305K BCL6 transfected into Raji and 293T cells. WT, wild-type.
We next asked whether the R305 residue of BCL6 was important for mediating the PRMT5-BCL6 interaction. To this end, we transfected plasmids encoding GFP-tagged wild-type or BCL6 R305K into Raji and 293T cells and performed co-IPs using PRMT5 or GFP antibodies. Indeed, the interaction of PRMT5 with BCL6 R305K was decreased in comparison with wild-type BCL6 (Figure 6D). Together, our findings suggest that PRMT5 contributes to the repressive activity of BCL6 through symmetric arginine dimethylation of BCL6 R305 and that this residue is important for the BCL6-PRMT5 interaction.
Inhibition of PRMT5 derepresses BCL6 target genes in DLBCL cells
Having observed that PRMT5 is necessary for BCL6-mediated repression, we next asked whether PRMT5 and BCL6 localize to the same genomic loci. To interrogate this, we transduced OCI-LY1 cells with HA-tagged PRMT5 and performed chromatin IP (ChIP) experiments for HA, BCL6, and its corepressor, SMRT. We observed enrichment of PRMT5 at several known BCL6 target genes, notably CCR6, BANK1 and CD69 but not at two negative control regions (Figure 7A). Treatment with the PRMT5-specific inhibitor GSK591 decreased occupancy of PRMT5, BCL6 and SMRT at these loci.
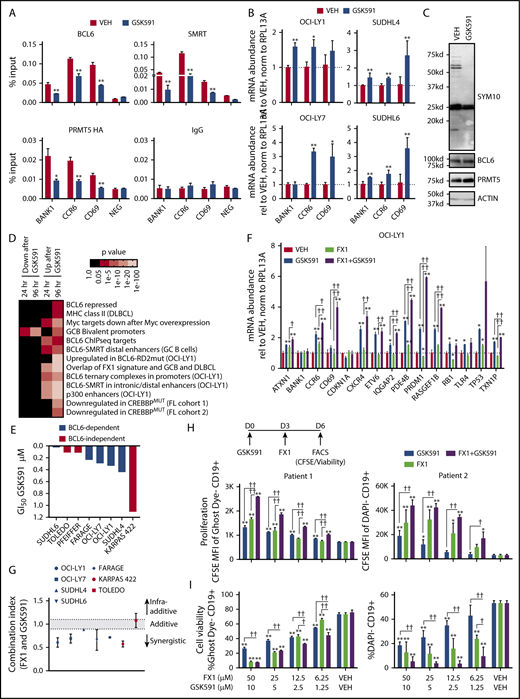
PRMT5 recruits BCL6 to its target genes to induce gene repression. (A) Enrichment of BCL6, SMRT, HA-tagged PRMT5, and IgG at BCL6 targets in OCI-LY1 cells treated with vehicle or 200 nM GSK591 for 72 hours. *P < .05; **P < .01. (B) Messenger RNA abundance of BCL6 target genes in OCI-LY1, OCI-LY7, SUDHL4, and SUDHL6 cells treated with vehicle or 200 nM GSK591 for 96 hours. *P < .05; **P < .01. (C) Immunoblot demonstrating inhibition of SDMA using the SYM10 antibody at 72 hours after treatment with 200 nM GSK591. IP demonstrating decrease in symmetric dimethylation of BCL6 shown in supplemental Figure 7A. (D) Pathway analysis of gene expression changes in SUDHL6 cells treated with vehicle or 200 nM GSK591 for 24 or 96 hours. The Fisher exact test was used to calculate enrichment P values for each gene set, and the Benjamini-Hochberg method was used for false discovery rate control. (E) GSK591 concentration that results in 50% growth inhibition (GI50) of BCL6-dependent and BCL6-independent DLBCL cell lines treated with vehicle or increasing concentrations of GSK591 for 6 days. Raw growth inhibition curves of GSK591 alone are shown in supplemental Figure 8A. (F) Messenger RNA abundance of BCL6 targets in OCI-LY1 cells with combined treatment of 25 μM FX1 and 200 nM GSK591 for 48 hours. *P < .05; **P < .01 relative to vehicle. †P < 0.05; ††P < .01 relative to each drug alone. (G) Combination indexes of the BCL6 inhibitor FX1 with GSK591 after treating cells with increasing concentrations of GSK591 for 6 days and FX1 for 2 days. Data are representative of 3 triplicates ± standard error of the mean (SEM). Raw growth inhibition curves of each drug alone and in combination are shown in supplemental Figure 8C. (H) Mean fluorescence intensity of carboxyfluorescein diacetate succinimidyl ester of live CD19+ (GhostDye− or DAPI−) human DLBCL samples on day 6 after cells were exposed to GSK591 on day 0 then treated with FX1 3 days later. Data representative of 3 triplicates ± SEM. *P < .05; **P < .01 relative to VEH. †P < 0.05; ††P < .01 relative to each drug alone. (I) Cell viability (GhostDye− or DAPI−) of CD19+ human DLBCL from panel H. Data are representative of 3 triplicates ± SEM. *P < .05; **P < .01 relative to vehicle. †P < .05; ††P < .01 relative to each drug alone. FACS, fluorescence-activated cell sorting; FL, follicular lymphoma; MFI, mean fluorescence intensity; MHC, major histocompatibility complex; mRNA, messenger RNA; VEH, vehicle.
PRMT5 recruits BCL6 to its target genes to induce gene repression. (A) Enrichment of BCL6, SMRT, HA-tagged PRMT5, and IgG at BCL6 targets in OCI-LY1 cells treated with vehicle or 200 nM GSK591 for 72 hours. *P < .05; **P < .01. (B) Messenger RNA abundance of BCL6 target genes in OCI-LY1, OCI-LY7, SUDHL4, and SUDHL6 cells treated with vehicle or 200 nM GSK591 for 96 hours. *P < .05; **P < .01. (C) Immunoblot demonstrating inhibition of SDMA using the SYM10 antibody at 72 hours after treatment with 200 nM GSK591. IP demonstrating decrease in symmetric dimethylation of BCL6 shown in supplemental Figure 7A. (D) Pathway analysis of gene expression changes in SUDHL6 cells treated with vehicle or 200 nM GSK591 for 24 or 96 hours. The Fisher exact test was used to calculate enrichment P values for each gene set, and the Benjamini-Hochberg method was used for false discovery rate control. (E) GSK591 concentration that results in 50% growth inhibition (GI50) of BCL6-dependent and BCL6-independent DLBCL cell lines treated with vehicle or increasing concentrations of GSK591 for 6 days. Raw growth inhibition curves of GSK591 alone are shown in supplemental Figure 8A. (F) Messenger RNA abundance of BCL6 targets in OCI-LY1 cells with combined treatment of 25 μM FX1 and 200 nM GSK591 for 48 hours. *P < .05; **P < .01 relative to vehicle. †P < 0.05; ††P < .01 relative to each drug alone. (G) Combination indexes of the BCL6 inhibitor FX1 with GSK591 after treating cells with increasing concentrations of GSK591 for 6 days and FX1 for 2 days. Data are representative of 3 triplicates ± standard error of the mean (SEM). Raw growth inhibition curves of each drug alone and in combination are shown in supplemental Figure 8C. (H) Mean fluorescence intensity of carboxyfluorescein diacetate succinimidyl ester of live CD19+ (GhostDye− or DAPI−) human DLBCL samples on day 6 after cells were exposed to GSK591 on day 0 then treated with FX1 3 days later. Data representative of 3 triplicates ± SEM. *P < .05; **P < .01 relative to VEH. †P < 0.05; ††P < .01 relative to each drug alone. (I) Cell viability (GhostDye− or DAPI−) of CD19+ human DLBCL from panel H. Data are representative of 3 triplicates ± SEM. *P < .05; **P < .01 relative to vehicle. †P < .05; ††P < .01 relative to each drug alone. FACS, fluorescence-activated cell sorting; FL, follicular lymphoma; MFI, mean fluorescence intensity; MHC, major histocompatibility complex; mRNA, messenger RNA; VEH, vehicle.
To determine whether inhibition of PRMT5 enzymatic activity affected expression of these genes, we treated the DLBCL cell lines OCI-LY1, OCI-LY7, SUDHL4, and SUDHL6 with 200 nM GSK591 and performed quantitative polymerase chain reaction to detect target gene transcript abundance. Loss of BCL6 function in DLBCL cells generally results in variable degrees of derepression of its direct target genes (usually in the two- to five-fold range).33,37 Accordingly, the PRMT5 inhibitor induced variable degrees of derepression of the BCL6 target genes CCR6, BANK1, and CD69 by quantitative polymerase chain reaction (Figure 7B). Importantly we confirmed that GSK591 suppressed total SDMA in these cells as shown by immunoblots with the SYM10 antibody and reduced levels of BCL6 SDMA (Figure 7C; supplemental Figure 7A). In contrast, GSK591 had no effect on BCL6 or PRMT5 protein levels (Figure 7C).
We next asked whether inhibiting PRMT5 would derepress BCL6 target genes on a broader level. To test this, we treated SUDHL6 cells with GSK591 and performed RNA-seq at 24 and 96 hours after treatment in biological triplicates. Notably, treatment with GSK591 induced derepression of canonical BCL6 target gene sets such as (1) genes induced by BCL6 siRNA (P = 9.43e-03), (2) genes induced by the BCL6 BTB domain inhibitor FX1 (P = 5.71e-95), (3) genes induced by loss of function of the BCL6 RD2 domain (P = 6.38e-69), and (4) genes regulated by BCL6 formation of corepressor complexes at gene promoters (P = 8.92e-20) and enhancers (P = 2.10e-43), respectively (Figure 7D; supplemental Table 1). As an alternative analysis approach, we performed gene set enrichment analysis, which also demonstrated significant enrichment (normalized enrichment score = 1.16, false discovery rate <0.05) of BCL6 repressed genes among genes induced by GSK591 treatment (supplemental Figure 7B). BCL6 normally antagonizes transcriptional activation by CREBBP and EP300 through its recruitment of SMRT/NCOR complexes and cooperates with EZH2 to repress GC bivalent genes. Notably, PRMT5 inhibitors induced derepression of known lymphoma CREBBP and EP300 target gene sets and induced derepression of GC bivalent genes (Figure 7D). Among gene sets downregulated after GSK591 treatment were those involved in cell-cycle control and MYC target genes, consistent with previously described roles for PRMT538,39 (supplemental Figure 7C).
PRMT5 inhibitors suppress DLBCL proliferation and enhance the effect of BCL6 inhibition
PRMT5 inhibitors were reported to suppress the proliferation of lymphoma cells,28 many of which are also dependent on BCL6. To confirm whether PRMT5 inhibitors could suppress the growth of DLBCL cells, we exposed a panel of BCL6-dependent and BCL6-independent cell lines to increasing doses of GSK591 (supplemental Figure 8A). PRMT5-targeted therapy suppressed the growth of most of these cell lines and did not manifest clear differences in growth inhibition (50% growth inhibition) among BCL6-dependent and BCL6-independent DLBCLs (Figure 7E). These results are consistent with the gene expression profiling results described above, which suggest that PRMT5 may impair other processes beyond BCL6 functions in DLBCL cells.
Our data suggest that PRMT5 inhibitors suppress BCL6 function through a different mechanism than currently existing BCL6 inhibitors, which block BCL6 recruitment of corepressors to its BTB domain.40 Therefore, we first asked whether adding PRMT5 inhibitors to BCL6 inhibitors leads to greater impairment of BCL6-mediated repression. We exposed DLBCL cell lines to a combination of GSK591 and the specific BCL6 inhibitor FX133 and found derepression of BCL6 target genes greater than that achieved with either compound alone (Figure 7F; supplemental Figure 8B). Moreover, combining GSK591 and the specific BCL6 inhibitor FX1 also yielded synergistic killing activity against BCL6-dependent DLBCL cells (Figure 7G; supplemental Figure 8C). Interestingly, we observe a synergistic effect on growth inhibition in a BCL6-positive BCL6-independent cell line (Karpas 422) with combination treatment of FX1 and GSK591, whereas an infra-additive response is observed in the BCL6-negative BCL6-independent cell line (Toledo) (Figure 7G). This is likely due to a relatively modest effect on growth inhibition with each single agent in Karpas 422 (supplemental Figure 8C). The combinatorial inhibition of BCL6 and PRMT5 therefore yields a greatly improved growth inhibition effect in Karpas 422 cells, resulting in a synergistic combination index. On the other hand, very little effect is observed in the Toledo cells with the BCL6 inhibitor alone. The PRMT5 inhibitor is quite effective at reducing growth as a single agent in Toledo cells. Therefore, when treated together, the PRMT5 and BCL6 inhibitors do not yield a further increase in growth inhibition in Toledo cells, resulting in only an infra-additive combination index (supplemental Figure 8C).
We wanted to confirm the combinatorial effects of GSK591 and FX1 on growth inhibition in primary DLBCL samples, as cell line models may not accurately represent primary human tumors. We obtained 4 viable primary DLBCL samples (2 GC-like and 2 non–GC-like) and grew them using 2 different methods. Samples from patient 1 grew successfully without any feeder layer; however, cells from patients 2, 3, and 4 had to be grown with a feeder layer of 40LB cells that expressed CD40 ligand and BAFF in an organoid system.41 Like other epigenetic-targeted agents, phenotypic effects of GSK591 occur after longer exposure to the drug. Therefore, cells were treated initially with GSK591 and then with FX1 72 hours later. We monitored the rate at which the proliferation dye carboxyfluorescein diacetate succinimidyl ester was diluted out using fluorescence-activated cell sorting. In patient 1, the combination of GSK591 and FX1 yielded significantly (P < .01) enhanced antiproliferative effects relative to each drug alone even at the lowest dose tested (6.25 μM FX1 and 1.25 μM GSK591; Figure 7H). While the combination of GSK591 and FX1 potently reduced cell viability at all concentrations, enhanced cell killing compared with each drug alone was only observed at lower concentrations, likely because of the strong antilymphoma effects of FX1 at higher concentrations (P < .01; Figure 7I). These results were confirmed in other patient samples (patients 2 and 3) (Figure 7H; supplemental Figure 8D-E). Patient 2 was more sensitive to the antiproliferative effects of FX1, so the drug combination had less profound effects (Figure 7H). Interestingly, PRMT5 and BCL6 inhibition did not have strong antiproliferative effects in patient 4. Instead, patient 4 had a strong decrease in cell viability with the drug combination relative to each agent alone at all concentrations tested (supplemental Figure 8D-E). Although these enhanced antilymphoma effects are likely multifactorial, our data suggest that targeting PRMT5 can enhance the effects of BCL6 inhibitors in antilymphoma regimens.
Discussion
The BCL6 protein is required for GC initiation, maintenance, and the development of high-affinity antigen-selected antibodies.1,2 This activity has been demonstrated to be mainly mediated by transcriptional repression through its N-terminal BTB/POZ and middle RD2 domains, which together affect the functions of BCL6 needed for the successful completion of the GC reaction.16,17 Although BCL6 interacts with a number of corepressor proteins, much remains to be understood regarding its molecular mechanism of action.
Several posttranslational modifications have been identified as regulating BCL6 function. Acetylation of BCL6 by EP300 disrupts its transcriptional repressor functions.42 Moreover, BCL6 phosphorylation and ubiquitinylation target the protein for proteosomal degradation.43 However, additional posttranslational modifications of BCL6 likely exist and play an important role in regulating BCL6 activity.
Herein, we identified the arginine methyltransferase PRMT5 as a novel interacting partner of BCL6. Our data demonstrate that BCL6 interacts with PRMT5 via its RD2 domain. Moreover, PRMT5 and its complex partner, MEP50, contribute to BCL6-mediated transcriptional repression by posttranslational arginine methylation of BCL6 residue R305. Mice with a GC B-cell–specific knockout of PRMT5 have decreased GC formation following immunization, similarly to mice with GC B-cell conditional deletion of BCL6. Furthermore, RNA-seq and ChIP experiments following treatment with a PRMT5 inhibitor demonstrate the derepression of many BCL6 target genes with a simultaneous decrease in BCL6 and SMRT enrichment at their promoters. CCR6 was among the targets where PRMT5 regulates BCL6 repressor activity. Interestingly, CCR6 knockout mice have an expanded GC B-cell phenotype reminiscent of that induced by BCL6 constitutive expression,44 and induction of CCR6 plays a critical role in memory B-cell function.45 These data allude to the importance of CCR6 being repressed by BCL6 during the GC reaction and derepressed for normal GC exit when BCL6 function is terminated. In DLBCL, derepression of CCR6 by targeted inhibition of PRMT5 and BCL6 might allow for the induction of GC exit programs and eventual cell death. These data underscore the importance of the cooperation between BCL6 and PRMT5 in governing BCL6-controlled transcriptional programs.
The effects of PRMT5 on BCL6 functions may go beyond its methylation of the R305 residue. In addition, while PRMT5 does not directly interact with the BTB/POZ domain of BCL6, it is found in complexes containing BCL6 BTB domain-interacting proteins (eg, NCOR and SMRT),36,46 and these proteins are important for mediating BCL6 repressive activity. Whether PRMT5 regulates BCL6’s BTB/POZ repressive ability via posttranslational modification of these repressive components is unknown and remains to be elucidated.
Our data also demonstrate that PRMT5 interacts with the C-terminal ZNF domain of BCL6. Unlike its interaction with the RD2 domain, this interaction did not mediate transcriptional repression, consistent with previous reports analyzing the function of the ZNF domain. However, PRMT5’s interaction with the ZNF domain may play a role in regulating BCL6 function in a nontranscriptional repressive capacity (ie, protein stability) and needs to be examined further.
Interestingly, PRMT5-targeted therapy caused growth inhibition in BCL6-independent DLBCL cell lines. However many studies have demonstrated pleotropic functions of PRMT5 in growth and survival. For example, PRMT5 has been shown to interact with and methylate other transcription factors such as E2F1, nuclear factor κB, and MYC.47-49 PRMT5 has also been shown to modulate the cell-cycle control proteins Rb and CDK4.50,51 Furthermore, PRMT5 regulates splicing by methylating Sm proteins.39,52 These proteins are all known to play important roles in the GC reaction and are implicated in B-cell lymphoma. Because of PRMT5’s multifaceted functions, we believe that BCL6-independent cell lines still require PRMT5 to regulate these other biological processes critical for their survival.
With our current work, we expand the role of PRMT5 in tumors to include facilitating the actions of BCL6, which is an oncogene in many tumor types other than lymphoma.40 Preclinical studies demonstrated that PRMT5 inhibition suppresses the growth of many tumors, including mantle cell lymphoma and leukemias, among others.26,53-55 Our studies demonstrate that inhibition of PRMT5 may provide additional suppression of BCL6 in tumors where BCL6 plays an important biological role. Hence the combination of PRMT5 and BCL6 inhibitors could be used as a general treatment strategy where inhibitors that affect 2 functions of a given oncogene are given together to promote tumor killing.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank GlaxoSmithKline for GSK591 and the Weill Cornell Epigenomics Core Facility for high throughput data processing.
I.S.L. is supported by the Dwoskin, Recio, and Anthony Rizzo Families Foundations, Jaime Erin Follicular Lymphoma Research Consortium, and University of Miami Sylvester Comprehensive Cancer Center. A.M.M. was supported by the National Institutes of Health, National Cancer Institute (R01CA155226 and P50 CA192937-01). F.L. was supported by an American Cancer Society institutional research grant.
Authorship
Contribution: X.L. designed and performed experiments and analyzed data; T.M.F. designed and performed experiments, analyzed data, and wrote the manuscript; C.L., A.M.M., and I.S.L. conceptualized the study, designed experiments, analyzed data, and wrote the manuscript; N.Y. and L.F. performed experiments and analyzed data; F.L. designed and generated reagents necessary for this study; M.D., J.M., Y.L., F.V., and V.M. performed experiments; H.G. analyzed data; G.I. provided primary patient samples; S.N. conceptualized the study, provided reagents, analyzed data, and wrote the manuscript; and all the authors read this manuscript and approved its content.
Correspondence: Izidore S. Lossos, Division of Hematology, University of Miami, Sylvester Comprehensive Cancer Center, 1475 NW 12th Ave (D8-4), Miami, FL 33136; e-mail: ilossos@med.miami.edu; and Ari M. Melnick, Division of Hematology/Oncology, Department of Medicine, Weill Cornell Medical College, 413 E 69th St, New York, NY 10021; e-mail: amm2014@med.cornell.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal