

Key Points
Deletion of both Rap1a and Rap1b impairs platelet production and abolishes platelet adhesion at sites of mechanical trauma.
Platelet RAP1 signaling is dispensable for vascular integrity during development and at sites of inflammation.

Abstract
RAP GTPases, important regulators of cellular adhesion, are abundant signaling molecules in the platelet/megakaryocytic lineage. However, mice lacking the predominant isoform, RAP1B, display a partial platelet integrin activation defect and have a normal platelet count, suggesting the existence of a RAP1-independent pathway to integrin activation in platelets and a negligible role for RAP GTPases in megakaryocyte biology. To determine the importance of individual RAP isoforms on platelet production and on platelet activation at sites of mechanical injury or vascular leakage, we generated mice with megakaryocyte-specific deletion (mKO) of Rap1a and/or Rap1b. Interestingly, Rap1a/b-mKO mice displayed a marked macrothrombocytopenia due to impaired proplatelet formation by megakaryocytes. In platelets, RAP isoforms had redundant and isoform-specific functions. Deletion of RAP1B, but not RAP1A, significantly reduced α-granule secretion and activation of the cytoskeleton regulator RAC1. Both isoforms significantly contributed to thromboxane A2 generation and the inside-out activation of platelet integrins. Combined deficiency of RAP1A and RAP1B markedly impaired platelet aggregation, spreading, and clot retraction. Consistently, thrombus formation in physiological flow conditions was abolished in Rap1a/b-mKO, but not Rap1a-mKO or Rap1b-mKO, platelets. Rap1a/b-mKO mice were strongly protected from experimental thrombosis and exhibited a severe defect in hemostasis after mechanical injury. Surprisingly, Rap1a/b-mKO platelets were indistinguishable from controls in their ability to prevent blood–lymphatic mixing during development and hemorrhage at sites of inflammation. In summary, our studies demonstrate an essential role for RAP1 signaling in platelet integrin activation and a critical role in platelet production. Although important for hemostatic/thrombotic plug formation, platelet RAP1 signaling is dispensable for vascular integrity during development and inflammation.
Introduction
Platelets are specialized cells generated from the cytoplasmic fragmentation of megakaryocytes to ensure the integrity of the vascular system upon mechanical injury or any other vascular breach, occurring, for instance, at sites of inflammation.1-3 Platelet stimulation triggers intracellular signaling cascades that promote cytoskeletal remodeling, secretion of granules, release of eicosanoids, and conversion of integrin receptors from a low- to a high-affinity state for their ligands (inside-out activation). Once active, integrins mediate platelet adhesion to the exposed extracellular matrix and aggregation to adjacent active platelets. Auto/paracrine agonists released from activated platelets, such as adenosine diphosphate (ADP) and thromboxane A2 (TxA2), locally perpetuate platelet activation and promote the formation of a stable shear-resistant hemostatic plug.4,5
The signaling machinery controlling these responses must be tightly regulated to prevent pathological thrombosis or bleeding. RAP GTPases (Ras-related proteins) are among the most abundant signaling proteins expressed in platelets.6-9 They are molecular switches that cycle between a GDP-bound “off” state and a GTP-bound “on” state, under the tight control of guanine-nucleotide exchange factors (GEFs) and GTPase-activating proteins (GAPs).10 Once GTP loaded, RAP GTPases undergo conformational changes that enable specific binding to effectors, which, in turn, control a wide range of biochemical events, most notably cell adhesion,11 cytoskeletal dynamics,12 and MAPK cascades.13
Vertebrates express 2 RAP1 isoforms and 3 RAP2 isoforms encoded by separate genes. Rap1a and Rap1b share a 95% sequence identity, suggesting that they may have redundant functions during cellular activation. However, there is also growing evidence that these 2 closely related isoforms differentially regulate cellular functions, such as NADPH oxidase activity14,15 and cadherin-mediated adhesion.16 In platelets, RAP1A and RAP1B are the most highly expressed members of the RAS superfamily.6-9 Our previous studies demonstrated that the activity state of RAP1 and platelet adhesiveness are controlled by the antagonistic balance between 2 upstream RAP regulators.17,18 In circulating platelets, RAP proteins are maintained inactive by the RAP-GAP RASA3.18 At sites of injury, when platelets are stimulated, the RAP-GEF CalDAG-GEFI mediates rapid RAP activation triggered by the near-immediate rise in cytosolic calcium concentrations17,19-21 ; however, this response is transient in nature, and RAP can quickly reverse back to the “off” state. Thus, signaling from secreted ADP via the P2Y12 receptor is critical to inactivate RASA3 and enable sustained RAP activation.18,22 Simultaneous loss of both RAP stimulatory pathways leads to marked defects in integrin activation,22 RAC1-mediated cytoskeletal dynamics,20,23 MAPK ERK signaling, and TxA2 generation.19
These studies provide indirect evidence that RAP signaling is critical for overall platelet activation and hemostatic plug formation. The only direct evidence supporting an important role for RAP1 signaling in platelet activation comes from studies in mice with systemic deletion of the predominant RAP1 isoform in platelets, RAP1B. Surprisingly, Rap1b−/− platelets displayed a rather mild platelet phenotype, including partially decreased agonist-induced fibrinogen binding and aggregation,24 reduced cytoskeleton-regulated responses (eg, secretion and spreading), and no defect in MAPK signaling.25 The role of RAP1A in the platelet hemostatic response has not been resolved, although Chrzanowska-Wodnicka et al reported unpublished observations that platelet aggregation is normal in RAP1A-null mice.24 Mice lacking RAP1A have impaired immune cell functions, which were not compensated for by RAP1B, suggesting that these isoforms may indeed have distinct roles.26 Interestingly, the peripheral platelet count and size were normal in Rap1a−/− and Rap1b−/− mice, suggesting a minimal role for RAP1 signaling in platelet production. This finding is surprising considering that RAP1 GTPases are highly expressed in platelet progenitor cells.27 However, very little28,29 has been done to investigate their role in megakaryocyte biology and platelet production.
In the present study, we characterized mice with targeted deletion of Rap1a and Rap1b selectively in the platelet/megakaryocyte lineage to investigate the specific contribution of the individual RAP isoforms to the regulation of platelet production, platelet activation, and hemostatic plug/thrombus formation. We demonstrate that RAP isoforms have redundant and isoform-specific functions in platelets and that they are essential for the regulation of hemostasis at sites of mechanical injury but are dispensable at sites of inflammation. Moreover, we identify a previously unrecognized role for RAP1 signaling in platelet production.
Methods
Mice
Generation of Rap1afl/flRap1bfl/fl 30 and Talin1fl/fl 31 mice has been described previously. Mice with megakaryocyte-specific deletion of (mKO) Rap1a (Rap1a-mKO), Rap1b (Rap1b-mKO), Rap1a/b (Rap1a/b-mKO), and their respective controls were generated by crossing Rap1afl/flRap1bfl/fl mice with C57BL/6-Tg(Pf4-Cre+) mice.32 Experimental procedures were approved by the Institutional Animal Care and Use Committees.
Megakaryocyte studies
Proplatelet formation was studied in mouse bone marrow–derived megakaryocytes, as described recently.18
Platelet studies
Platelet count, size, and lifespan, surface receptor expression, integrin αIIbβ3 activation, and α-granule secretion were determined, as described recently.18 Integrin β1 activation was determined by measuring the binding of an antibody directed against the active form of murine β1 (clone 9EG7).33 Aggregometry and RAP2 pull-down assays were performed as previously described.23 Ex vivo flow studies were performed in a collagen-coated microfluidic chamber, as previously described.22
Thrombosis and hemostasis studies
Ferric chloride (FeCl3)-induced thrombosis to the common carotid artery was performed as described previously.34 Tail bleeding time and blood loss volume were measured as previously described.22 Hemostatic plug formation and bleeding time upon laser-induced injury to the saphenous vein were studied as described recently.35
Vascular integrity studies
The contribution of platelets to vascular integrity at sites of inflammation was determined by challenging mice with the reverse passive Arthus (rpA) reaction, as described previously.36 Preparation and immunohistochemistry of embryos, at approximately embryonic day 16.5 (E16.5), to assess blood–lymphatic mixing (BLM) was performed as described recently.18
Statistics
Results are reported as mean ± standard error of the mean (SEM), and statistical significance was assessed by ANOVA, unless otherwise indicated. P < .05 was considered significant.
Additional information is provided in supplemental Methods, available on the Blood Web site.
Results
Combined deficiency of RAP1A and RAP1B in the platelet/megakaryocytic lineage leads to macrothrombocytopenia
Systemic deletion of both RAP1 isoforms is not viable in mice.37 To investigate the respective roles of RAP1A and RAP1B in platelet biology, we crossed floxed Rap1a and Rap1b mice30 with C57BL/6-Tg(Pf4-Cre) mice,32 in which Cre recombinase expression is driven by the Pf4 promoter. Megakaryocyte-specific Rap1a/b double-knockout mice (Rap1a/b-mKO, Rap1afl/flRap1bfl/flPf4-Cre+) were viable and fertile and appeared overall healthy, with no spontaneous bleeding seen. Mice were routinely genotyped to detect the floxed Rap1 alleles and the Cre recombinase gene (supplemental Figure 1A). Western blot analysis confirmed the complete ablation of both RAP1 isoforms in Rap1a/b-mKO platelets and deletion of RAP1A or RAP1B in Rap1a-mKO (Rap1afl/flRap1b+/+Pf4-Cre+) and Rap1b-mKO (Rap1a+/+Rap1bfl/flPf4-Cre+) mice, respectively (supplemental Figure 1B). Platelet count and platelet size were normal in Rap1a-mKO mice (data not shown). Rap1b-mKO mice exhibited a normal platelet count and a slight increase in platelet size (Figure 1A-B). Concomitant deletion of 1 allele of Rap1a led to a further increase in platelet size (Figure 1B). Rap1a/b-mKO mice displayed an ∼40% reduction in platelet count and an ∼50% increase in platelet size. The marked thrombocytopenia observed in Rap1a/b-mKO mice is likely the result of impaired platelet production, because we did not observe a reduction, but rather a slight increase, in the half-life of Rap1a/b-mKO platelets in circulation (Figure 1C) and a significant defect in proplatelet formation by megakaryocytes ex vivo (Figure 1D-E).
![Figure 1. Megakaryocyte-specific deletion of the Rap1a and Rap1b genes leads to macrothrombocytopenia. Flow cytometric analysis of the peripheral platelet count (A) and platelet size (forward scatter height [FSH]) (B) in mice lacking 1 or both alleles of the Rap1a and Rap1b genes in the platelet/megakaryocyte lineage, as indicated below each bar (n = 12). (C) Platelet lifespan assay. Endogenous platelets were labeled by infusion of Alexa Fluor 488–conjugated antibody to GPIX, and the percentage of GPIX-labeled platelets remaining was determined by flow cytometry every 24 hours. (D) In vitro proplatelet (PP) formation in bone marrow–derived megakaryocytes. (E) Representative images of megakaryocytes for PP-formation assay, acquired on an Olympus IX-81 wide-field microscope with a Photometrics CoolSnap HQ2 camera. *P < .05, **P < .01, ***P < .001. ns, not significant.](https://ash.silverchair-cdn.com/ash/content_public/journal/blood/132/18/10.1182_blood-2018-03-838714/4/m_blood838714f1.png?Expires=1769137412&Signature=wK5VqgrOxicAXigPJIecjUuIYptcno49lq9~yyNdCX17UPqLooQ3~Oo1YIuCjx9ntyRme3twsd53peLb5kUaC-uwYmHQkwGYgg5mts-EyV5yP-JRPmLuK5bg8ARPXUJC~sax0aKckux6KcgMSvfmkEb9nrzZ5f~mYyfib~1fPGjxjj7J7~nOx3wjmUSYvCy76jrT~Rs9xVBHPXItktrsudeMaJ9T0YKqZyqbKfDj7arcFhP95c1CPcQiKCZDxkSwsQu8YEqJWen7Hx0yvPZgjOkdy6Itx7~WzP9Vawkm4weXcMpviyKK~dCnnmtVrxkp~zZ1BY6UGYhiBEL6kThGdg__&Key-Pair-Id=APKAIE5G5CRDK6RD3PGA)
Megakaryocyte-specific deletion of the Rap1a and Rap1b genes leads to macrothrombocytopenia. Flow cytometric analysis of the peripheral platelet count (A) and platelet size (forward scatter height [FSH]) (B) in mice lacking 1 or both alleles of the Rap1a and Rap1b genes in the platelet/megakaryocyte lineage, as indicated below each bar (n = 12). (C) Platelet lifespan assay. Endogenous platelets were labeled by infusion of Alexa Fluor 488–conjugated antibody to GPIX, and the percentage of GPIX-labeled platelets remaining was determined by flow cytometry every 24 hours. (D) In vitro proplatelet (PP) formation in bone marrow–derived megakaryocytes. (E) Representative images of megakaryocytes for PP-formation assay, acquired on an Olympus IX-81 wide-field microscope with a Photometrics CoolSnap HQ2 camera. *P < .05, **P < .01, ***P < .001. ns, not significant.
Megakaryocyte-specific deletion of the Rap1a and Rap1b genes leads to macrothrombocytopenia. Flow cytometric analysis of the peripheral platelet count (A) and platelet size (forward scatter height [FSH]) (B) in mice lacking 1 or both alleles of the Rap1a and Rap1b genes in the platelet/megakaryocyte lineage, as indicated below each bar (n = 12). (C) Platelet lifespan assay. Endogenous platelets were labeled by infusion of Alexa Fluor 488–conjugated antibody to GPIX, and the percentage of GPIX-labeled platelets remaining was determined by flow cytometry every 24 hours. (D) In vitro proplatelet (PP) formation in bone marrow–derived megakaryocytes. (E) Representative images of megakaryocytes for PP-formation assay, acquired on an Olympus IX-81 wide-field microscope with a Photometrics CoolSnap HQ2 camera. *P < .05, **P < .01, ***P < .001. ns, not significant.
Signaling by RAP1A and RAP1B contributes to platelet integrin activation
Given the documented role of RAP1 signaling in cell adhesion,11 we next investigated the role of both RAP1 isoforms in integrin activation. Deletion of both isoforms did not impair the surface expression of important adhesion receptors, including αIIbβ3 and β1 integrins (supplemental Figure 1C). Agonist-induced activation of platelet integrins was first evaluated by flow cytometry using conformation-sensitive antibodies against activated αIIbβ3 (Figure 2A) and β1 integrins (Figure 2B). Consistent with studies in conventional Rap1b-knockout mice,24 integrin activation was partially reduced in Rap1b-mKO platelets activated with various agonists. A more modest, but significant, defect in integrin activation was observed in Rap1a-mKO platelets, revealing a previously unrecognized role for this isoform in platelet inside-out signaling. Importantly, combined deficiency of RAP1A and RAP1B led to an almost complete (80%-90%) inhibition of αIIbβ3 and β1 integrin activation, even at high doses of strong agonists.

RAP1A and RAP1B contribute to platelet integrin activation. Flow cytometric analysis of integrin αIIbβ3 activation (binding of Jon/A-PE; clone Leo.H4; Emfret Analytics) (A) and integrin β1 activation (binding of 9EG7–fluorescein isothiocyanate; BD Biosciences) (B) in response to increasing concentrations of thrombin, the GPVI-agonist convulxin, or the combination of ADP and the TxA2 analog U46619. Data shown are mean fluorescence intensity (MFI) ± SEM of control (Rap1afl/flRap1bfl/flPf4-Cre−), Rap1a-mKO (Rap1afl/flRap1bwt/wtPf4-Cre+), Rap1b-mKO (Rap1awt/wtRap1bfl/flPf4-Cre+), and Rap1a/b-mKO (Rap1afl/flRap1bfl/flPf4-Cre+) platelets (n = 6). *P < .05, **P < .01, ***P < .001. ns, not significant.
RAP1A and RAP1B contribute to platelet integrin activation. Flow cytometric analysis of integrin αIIbβ3 activation (binding of Jon/A-PE; clone Leo.H4; Emfret Analytics) (A) and integrin β1 activation (binding of 9EG7–fluorescein isothiocyanate; BD Biosciences) (B) in response to increasing concentrations of thrombin, the GPVI-agonist convulxin, or the combination of ADP and the TxA2 analog U46619. Data shown are mean fluorescence intensity (MFI) ± SEM of control (Rap1afl/flRap1bfl/flPf4-Cre−), Rap1a-mKO (Rap1afl/flRap1bwt/wtPf4-Cre+), Rap1b-mKO (Rap1awt/wtRap1bfl/flPf4-Cre+), and Rap1a/b-mKO (Rap1afl/flRap1bfl/flPf4-Cre+) platelets (n = 6). *P < .05, **P < .01, ***P < .001. ns, not significant.
Limited integrin activation, mediated by TALIN1, in platelets lacking both RAP1 isoforms is not mediated by RAP2 GTPases
TALIN binding to the integrin β tail is the final common step that mediates integrin activation. Elegant studies in heterologous cell lines stably expressing αIIbβ338-40 demonstrated that RAP1 mediates integrin activation by targeting the cytoskeletal protein TALIN to the plasma membrane. To test whether RAP1 is essential for TALIN-mediated integrin activation, we next compared Rap1a/b-mKO and Talin1-mKO platelets.31 Consistent with previous reports,31,41,42 agonist-induced JON/A-PE binding and aggregation response were virtually abolished in Talin1-mKO platelets (Figure 3A-B). Compared with Talin1-mKO platelets, Rap1a/b-mKO platelets exhibited a small, but significant, increase in JON/A-PE binding and delayed aggregation in response to high doses of agonists (with the exception of ADP). The maximum extent of aggregation was always reduced in Rap1a/b-mKO platelets compared with control platelets, and aggregates of Rap1a/b-mKO platelets appeared smaller by visual inspection (data not shown). Residual aggregation of Rap1a/b-mKO platelets was inhibited by preincubation with a function blocking antibody to murine αIIbβ3 (Figure 3B). Thus, although Talin1-mKO platelets were intrinsically incapable of activating αIIbβ3 integrin, a limited amount of integrin-mediated aggregation was observed in platelets lacking RAP1A and RAP1B.

Limited TALIN1-mediated integrin activation in platelets lacking both RAP1 isoforms is not mediated by RAP2 GTPase. (A) Flow cytometric analysis of αIIbβ3 integrin activation (JON/A-PE antibody binding) in Rap1a/b-mKO (Rap1afl/flRap1bfl/flPf4-Cre+) and Talin1-mKO (Talin1fl/flPf4-Cre+) platelets activated with high dose thrombin (HD Thr) or high dose convulxin (HD Cvx) (n = 5). (B) Aggregation response of Rap1a/b-mKO and Talin1-mKO platelets (representative of 4 independent experiments). For high-dose Par4p and convulxin (Cvx) aggregation, the residual aggregation response of Rap1a/b-mKO platelets was completely abolished by preincubation with 30 μg/mL of the αIIbβ3 blocking antibody Leo.H4. (C) Rap2-GTP pull-down assay. Washed platelets stimulated (S) in standard aggregometry with 0, 0.4 μg/mL, or 0.8 μg/mL the GPVI-agonist Cvx were lysed (L) after 10 minutes to measure the levels of active RAP2 by pull-down assay (n = 3). Total RAP2 was determined as loading control. (D) Platelet spreading on fibrinogen. Platelets were incubated in fibrinogen-coated wells and activated with ADP (100 µM), and the extent of platelet spreading was determined after 45 minutes. Representative images of phalloidin-stained platelets are shown (n = 3-5). Scale bars represent 10 μm. (E) Clot-contraction assay. Washed platelets from the indicated knockout mice were added to human platelet-poor plasma in ACD containing 5 mM Ca2+ and 0.2 U/mL thrombin in siliconized cuvettes (n = 3). Representative images of clots at time 0 and 120 minutes are shown (upper panel). The “no platelets” sample contained all components with the exception of washed platelets. *P < .05, **P < .01, ***P < .001. ns, not significant.
Limited TALIN1-mediated integrin activation in platelets lacking both RAP1 isoforms is not mediated by RAP2 GTPase. (A) Flow cytometric analysis of αIIbβ3 integrin activation (JON/A-PE antibody binding) in Rap1a/b-mKO (Rap1afl/flRap1bfl/flPf4-Cre+) and Talin1-mKO (Talin1fl/flPf4-Cre+) platelets activated with high dose thrombin (HD Thr) or high dose convulxin (HD Cvx) (n = 5). (B) Aggregation response of Rap1a/b-mKO and Talin1-mKO platelets (representative of 4 independent experiments). For high-dose Par4p and convulxin (Cvx) aggregation, the residual aggregation response of Rap1a/b-mKO platelets was completely abolished by preincubation with 30 μg/mL of the αIIbβ3 blocking antibody Leo.H4. (C) Rap2-GTP pull-down assay. Washed platelets stimulated (S) in standard aggregometry with 0, 0.4 μg/mL, or 0.8 μg/mL the GPVI-agonist Cvx were lysed (L) after 10 minutes to measure the levels of active RAP2 by pull-down assay (n = 3). Total RAP2 was determined as loading control. (D) Platelet spreading on fibrinogen. Platelets were incubated in fibrinogen-coated wells and activated with ADP (100 µM), and the extent of platelet spreading was determined after 45 minutes. Representative images of phalloidin-stained platelets are shown (n = 3-5). Scale bars represent 10 μm. (E) Clot-contraction assay. Washed platelets from the indicated knockout mice were added to human platelet-poor plasma in ACD containing 5 mM Ca2+ and 0.2 U/mL thrombin in siliconized cuvettes (n = 3). Representative images of clots at time 0 and 120 minutes are shown (upper panel). The “no platelets” sample contained all components with the exception of washed platelets. *P < .05, **P < .01, ***P < .001. ns, not significant.
Platelets also express all RAP2 isoforms, RAP2A, RAP2B, and RAP2C,6-8 which are under the control of the same GEF23 and are 70% homologous to RAP1.43 Thus, we investigated whether RAP2 GTPases could be responsible for the RAP1-independent TALIN1-dependent integrin activation elicited by high doses of agonists in Rap1a/b-mKO platelets. We stimulated Rap1a/b-mKO, Talin1-mKO, and control platelets for 5 minutes with 2 distinct concentrations of agonist, above or below the aggregating threshold of Rap1a/b-mKO platelets, and then measured RAP2-GTP levels in these samples (Figure 3C). We detected comparable levels of active RAP2 in platelets from all genotypes. Moreover, we found no correlation between RAP2 activation and the level of platelet aggregation reached by these platelets (ie, RAP2 was activated in aggregating and nonaggregating platelets). Thus, it is unlikely that RAP2 signaling mediates integrin activation in platelets deficient in RAP1.
Loss of RAP1A/B isoforms impairs platelet spreading and clot contraction
Following inside-out activation of αIIbβ3 integrin, ligand binding triggers outside-in signaling, which plays an essential role in platelet spreading and clot contraction by linking integrin activation with cytoskeletal mechanical forces.44 Previous work has reported that fibrinogen binding induces RAP1 GTP loading25 and that Rap1b−/− platelets exhibit defects in spreading24,25 and clot contraction.25 To analyze the ability of our RAP-mutant platelets to spread, cells were added to fibrinogen-coated wells and activated with ADP at 37°C, and the platelet area and the percentage of fully spread platelets were quantified. Under these conditions, ∼85% to 90% of control platelets were fully spread. Rap1a-mKO and Rap1b-mKO platelets spread normally compared with controls, whereas Rap1a/b-mKO platelets had an ∼50% reduction in platelet area and displayed an abnormal nonspread morphology (Figure 3D). Clot contraction was also partially impaired in Rap1a/b-mKO platelets (Figure 3E). In the platelet-spreading and clot-contraction assays, the defect in Rap1a/b-mKO platelets was less severe than that of Talin1-mKO platelets.
Specific RAP isoforms differentially regulate the release of second-wave mediators
Deficiency in CalDAG-GEFI affects platelet integrin activation, as well as the release of second-wave mediators,17,19,20,23 which is crucial for full platelet activation and hemostatic plug formation. Therefore, we next investigated the contribution of RAP1 isoforms to granule secretion and TxA2 generation. As previously described for conventional Rap1b-KO mice,25 RAP1B deficiency in platelets impaired α-granule secretion, in response to GPVI, but not PAR4, receptor stimulation (Figure 4A-B). In contrast, deletion of Rap1A alone did not affect P-selectin exposure, and Rap1a/b-mKO platelets displayed the same secretion phenotype as Rap1b-mKO platelets. Our previous studies suggested important cross talk between RAP1 and RAC1 in the regulation of actin dynamics and granule secretion in platelets activated via GPVI.23 Consistently, Rap1b-mKO and Rap1a/b-mKO platelets, but not Rap1a-mKO platelets, failed to activate RAC1 in response to convulxin stimulation at early time points (Figure 4C-D). RAC1-GTP levels were also significantly reduced in Rap1a/b-mKO platelets at the later time point after addition of the agonist.
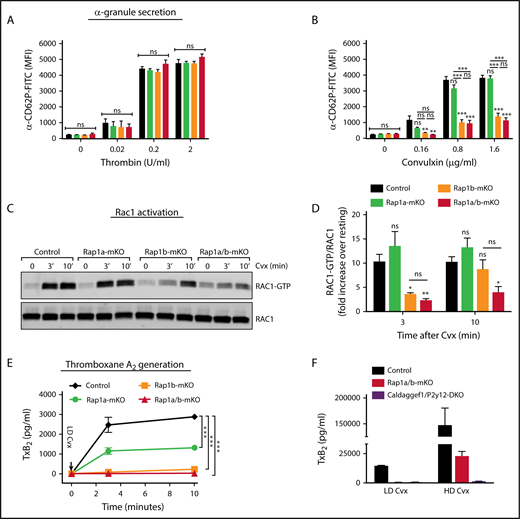
Specific RAP isoforms regulate granule secretion, RAC1 activation, and TxA2generation. Dose response studies of thrombin-induced (A) or convulxin-induced (B) P-selectin exposure (anti-CD62P binding by flow cytometry; clone RB40.34; BD Biosciences). Data shown are mean fluorescence intensity (MFI) ± SEM of control (Rap1afl/flRap1bfl/flPf4-Cre−; black), Rap1a-mKO (Rap1afl/flRap1bwt/wtPf4-Cre+; green), Rap1b-mKO (Rap1awt/wtRap1bfl/flPf4-Cre+; orange), and Rap1a/b-mKO (Rap1afl/flRap1bfl/flPf4-Cre+; red) platelets (n = 6). (C-D) RAC1-GTP pull-down assay. (C) RAC1 activation was determined by pull-down assay in platelets of the indicated genotype stimulated for 0, 3, or 10 minutes with convulxin (Cvx). Total RAC1 was determined as loading control. Western blot images are representative of 3 independent experiments. (D) The ratio of RAC1-GTP over RAC1 band intensity for the 3 experiments is shown as fold increase over resting. (E-F) TxB2-generation assay. (E) Washed platelets from control, Rap1a-mKO, Rap1b-mKO, and Rap1a/b-mKO mice were stimulated in standard aggregometry (data not shown) with low doses of Cvx (LD Cvx; 0.16 µg/mL). After 0, 3, and 10 minutes of stimulation, samples were withdrawn to measure the levels of TxB2, the stable product of TxA2. (F) In similar experimental conditions, Rap1a/b-mKO and Caldaggef1−/−P2y12−/− platelets were stimulated for 10 minutes with low-dose Cvx (LD Cvx; 0.16 µg/mL) or high-dose Cvx (HD Cvx; 1.6 µg/mL). *P < .05, **P < .01, ***P < .001. ns, not significant.
Specific RAP isoforms regulate granule secretion, RAC1 activation, and TxA2generation. Dose response studies of thrombin-induced (A) or convulxin-induced (B) P-selectin exposure (anti-CD62P binding by flow cytometry; clone RB40.34; BD Biosciences). Data shown are mean fluorescence intensity (MFI) ± SEM of control (Rap1afl/flRap1bfl/flPf4-Cre−; black), Rap1a-mKO (Rap1afl/flRap1bwt/wtPf4-Cre+; green), Rap1b-mKO (Rap1awt/wtRap1bfl/flPf4-Cre+; orange), and Rap1a/b-mKO (Rap1afl/flRap1bfl/flPf4-Cre+; red) platelets (n = 6). (C-D) RAC1-GTP pull-down assay. (C) RAC1 activation was determined by pull-down assay in platelets of the indicated genotype stimulated for 0, 3, or 10 minutes with convulxin (Cvx). Total RAC1 was determined as loading control. Western blot images are representative of 3 independent experiments. (D) The ratio of RAC1-GTP over RAC1 band intensity for the 3 experiments is shown as fold increase over resting. (E-F) TxB2-generation assay. (E) Washed platelets from control, Rap1a-mKO, Rap1b-mKO, and Rap1a/b-mKO mice were stimulated in standard aggregometry (data not shown) with low doses of Cvx (LD Cvx; 0.16 µg/mL). After 0, 3, and 10 minutes of stimulation, samples were withdrawn to measure the levels of TxB2, the stable product of TxA2. (F) In similar experimental conditions, Rap1a/b-mKO and Caldaggef1−/−P2y12−/− platelets were stimulated for 10 minutes with low-dose Cvx (LD Cvx; 0.16 µg/mL) or high-dose Cvx (HD Cvx; 1.6 µg/mL). *P < .05, **P < .01, ***P < .001. ns, not significant.
Our previous studies also demonstrated impaired TxA2 generation in platelets lacking CalDAG-GEFI.19 Consistently, we found that both RAP1 isoforms contributed to TxA2 generation, with Rap1b-mKO platelets displaying a phenotype almost comparable to that of the Rap1a/b-mKO platelets and RAP1A playing a minor role (Figure 4E). However, although simultaneous loss of both RAP stimulatory pathways (Caldaggef1−/−P2y12−/−) completely blocked GPVI-mediated TxA2 generation, the defect in this response was partially overcome in Rap1a/b-mKO platelets stimulated with a high concentration of agonist (Figure 4F).
RAP1A and RAP1B are critical for platelet adhesion under shear conditions and the formation of thrombotic and hemostatic plugs
To assess the contribution of RAP1A and RAP1B to thrombus formation under physiological shear stress conditions, we next studied platelet adhesion to collagen ex vivo in a microfluidics chamber assay (Figure 5A). Under these conditions, combined deficiency of RAP1A and RAP1B strongly inhibited platelet adhesion to collagen and completely prevented platelet–platelet cohesion (Figure 5B-C). Expression of a single Rap1a allele significantly improved platelet adhesion compared with Rap1a/b-mKO platelets, and it allowed for the formation of few very small thrombi. Platelet adhesion and platelet–platelet cohesion were further improved in blood from mice expressing only a single copy of Rap1b, confirming that RAP1B is more effective than RAP1A in supporting integrin-mediated platelet adhesion.
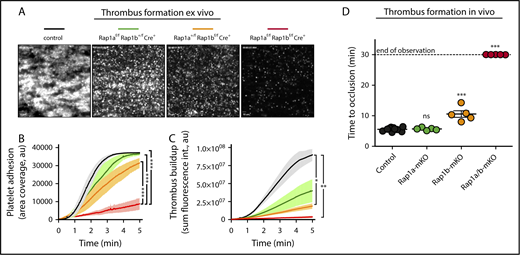
RAP1A and RAP1B are critical for platelet adhesion and thrombus formation under shear conditions ex vivo and in vivo. (A-C) Platelet adhesion to fibrillar collagen ex vivo. Anticoagulated whole blood from control (Rap1afl/flRap1bfl/flPf4-Cre-; black), Rap1afl/flRap1b+/flPf4-Cre+ (green), Rap1a+/flRap1bfl/flPf4-Cre+ (orange), or Rap1afl/flRap1bfl/flPf4-Cre+ (red) mice was perfused over fibrillar collagen type I (200 μg/mL) at venous shear rates (400−s). Adhesion of platelets was monitored continuously. Shown are representative images after 5 minutes of perfusion of Alexa Fluor 488–α-GPIX–labeled whole blood obtained using a Nikon TE300 (Nikon, Tokyo, Japan) microscope equipped with a QImaging Retiga EXi camera (Qimaging, Surrey, BC, Canada) (A), platelet adhesion determined as the area of adhesion coverage ± SEM (B), and thrombus buildup quantified as sum fluorescence intensity ± SEM (C). Analyses were performed using SlideBook 5.0 software (Intelligent Imaging Innovations, Denver, CO). Scale bars in A represent 10 μm. (D) FeCl3-induced thrombosis in the carotid artery. Data shown are the scatter dot plot (line at median) of the time of occlusion (minutes). Blood flow velocity was monitored for 30 minutes with a 0.5-mm Doppler flow probe connected to a Transonic TS420 Flow Module (Transonic, Ithaca, NY); time to occlusion was recorded when blood velocity reached 25% of baseline velocity. All Rap1a/b-mKO vessels never dropped below 25% of baseline flow; thus, they were assigned a time of 30 minutes. *P < .05, **P < .01, ***P < .001. ns, not significant.
RAP1A and RAP1B are critical for platelet adhesion and thrombus formation under shear conditions ex vivo and in vivo. (A-C) Platelet adhesion to fibrillar collagen ex vivo. Anticoagulated whole blood from control (Rap1afl/flRap1bfl/flPf4-Cre-; black), Rap1afl/flRap1b+/flPf4-Cre+ (green), Rap1a+/flRap1bfl/flPf4-Cre+ (orange), or Rap1afl/flRap1bfl/flPf4-Cre+ (red) mice was perfused over fibrillar collagen type I (200 μg/mL) at venous shear rates (400−s). Adhesion of platelets was monitored continuously. Shown are representative images after 5 minutes of perfusion of Alexa Fluor 488–α-GPIX–labeled whole blood obtained using a Nikon TE300 (Nikon, Tokyo, Japan) microscope equipped with a QImaging Retiga EXi camera (Qimaging, Surrey, BC, Canada) (A), platelet adhesion determined as the area of adhesion coverage ± SEM (B), and thrombus buildup quantified as sum fluorescence intensity ± SEM (C). Analyses were performed using SlideBook 5.0 software (Intelligent Imaging Innovations, Denver, CO). Scale bars in A represent 10 μm. (D) FeCl3-induced thrombosis in the carotid artery. Data shown are the scatter dot plot (line at median) of the time of occlusion (minutes). Blood flow velocity was monitored for 30 minutes with a 0.5-mm Doppler flow probe connected to a Transonic TS420 Flow Module (Transonic, Ithaca, NY); time to occlusion was recorded when blood velocity reached 25% of baseline velocity. All Rap1a/b-mKO vessels never dropped below 25% of baseline flow; thus, they were assigned a time of 30 minutes. *P < .05, **P < .01, ***P < .001. ns, not significant.
Consistent with the ex vivo studies, we observed severely reduced thrombus formation after FeCl3 injury to the carotid artery in Rap1a/b-mKO mice (Figure 5D). No defect in arterial thrombosis was observed in Rap1a-mKO mice, whereas a small, but significant, prolongation in the occlusion time was observed in Rap1b-mKO mice. Thus, RAP1A and RAP1B contribute to platelet adhesion and thrombus formation under flow conditions ex vivo and in vivo.
To determine the contribution of individual RAP1 isoforms to hemostatic plug formation in vivo, we challenged our RAP1-knockout mice in the standard tail-transection model (Figure 6A-B) and our recently developed laser injury–induced saphenous vein hemostasis model35 (Figure 6C-D). Although expression of a single allele of Rap1b was sufficient to promote hemostasis in both models, a significant prolongation in the bleeding time and an increase in the blood loss volume from the severed tail were observed in mice expressing 1 allele of Rap1a and in Rap1a/b-mKO mice (Figure 6A-B). Interestingly, only Rap1a/b-mKO mice showed a significant increase in the bleeding time after laser injury, comparable to that observed in Talin1-mKO mice (Figure 6C). Compared with the tail-transection model, laser-induced injuries to the endothelial lining are small (∼50 µm in diameter). Platelets expressing 1 allele of Rap1b were able to plug these small lesions, but platelet plugs were significantly smaller than in control mice (Figure 6D; supplemental Videos 1-4). In conclusion, these ex vivo and in vivo studies demonstrate that signaling by RAP1A can partially compensate for loss of RAP1B during hemostatic and thrombotic plug formation.

Platelet RAP1 signaling is critical for hemostasis at sites of injury. Determination of hemostasis using tail clip (A-B) or saphenous vein laser injury (C-D) model in control (Rap1afl/flRap1bfl/flPf4-Cre−; black bars; black line), Rap1afl/flRap1b+/flPf4-Cre+ (green bars; green line), Rap1a+/flRap1bfl/flPf4-Cre+ (orange bars; orange line), and Rap1a/b-mKO (Rap1afl/flRap1bfl/flPf4-Cre+; red bars; red line) mice. Tail bleeding time (A) and blood loss volume (B) were determined following tail clipping. (C) Repeated laser injuries were made to the saphenous vein using an Ablate! photoablation system equipped with an attenuable 532-nm pulse laser (Intelligent Imaging Innovations, Denver, CO). Bleeding time was assessed as time (seconds) to stable hemostatic plug formation (no leakage of blood for >60 seconds) within the laser injury–induced vascular lesion. Talin1-mKO mice (Talin1fl/flPf4-Cre+; blue bar), which bleed for the entire observation period, are shown for comparison. (D) Platelet accumulation was recorded using a Zeiss Axio Examiner Z1 microscope (Intelligent Imaging Innovations) equipped with a 20×/1 numerical aperture water immersion objective lens and determined as sum fluorescence intensity (SFI) of GPIX-labeled platelets ± SEM at the site of injury over time; data were analyzed using SlideBook 5.0 software (Intelligent Imaging Innovations). *P < .05, **P < .01, ***P < .001. ns, not significant.
Platelet RAP1 signaling is critical for hemostasis at sites of injury. Determination of hemostasis using tail clip (A-B) or saphenous vein laser injury (C-D) model in control (Rap1afl/flRap1bfl/flPf4-Cre−; black bars; black line), Rap1afl/flRap1b+/flPf4-Cre+ (green bars; green line), Rap1a+/flRap1bfl/flPf4-Cre+ (orange bars; orange line), and Rap1a/b-mKO (Rap1afl/flRap1bfl/flPf4-Cre+; red bars; red line) mice. Tail bleeding time (A) and blood loss volume (B) were determined following tail clipping. (C) Repeated laser injuries were made to the saphenous vein using an Ablate! photoablation system equipped with an attenuable 532-nm pulse laser (Intelligent Imaging Innovations, Denver, CO). Bleeding time was assessed as time (seconds) to stable hemostatic plug formation (no leakage of blood for >60 seconds) within the laser injury–induced vascular lesion. Talin1-mKO mice (Talin1fl/flPf4-Cre+; blue bar), which bleed for the entire observation period, are shown for comparison. (D) Platelet accumulation was recorded using a Zeiss Axio Examiner Z1 microscope (Intelligent Imaging Innovations) equipped with a 20×/1 numerical aperture water immersion objective lens and determined as sum fluorescence intensity (SFI) of GPIX-labeled platelets ± SEM at the site of injury over time; data were analyzed using SlideBook 5.0 software (Intelligent Imaging Innovations). *P < .05, **P < .01, ***P < .001. ns, not significant.
RAP1 GTPases play a minor role for embryonic vascular development and vascular integrity at sites of inflammation in the adult
In addition to their classical role in the prevention of excessive blood loss at sites of mechanical injury, platelets safeguard vascular integrity during development and at sites of inflammation.45-47 To test whether platelet RAP1-TALIN signaling is important during development, we studied embryos from Rap1a/b-mKO and Talin1-mKO mice for signs of BLM. Compared with controls, Rap1a/b-mKO embryos (∼E16.5) showed very mild signs of BLM, as shown by macroscopic observation of whole embryos and immunostaining of embryo sections (Figure 7A). Markedly more severe BLM was observed in some Talin1-mKO embryos, whereas others showed only mild defects. Similar to our observations in the developing embryos, Talin1-mKO mice, but not Rap1a/b-mKO mice, exhibited significant hemorrhage in a model of immune complex–mediated inflammation in the skin (rpA reaction) (Figure 7B). However, hemorrhage at sites of inflammation in Talin1-mKO mice was significantly less than in control mice rendered thrombocytopenic (<2% platelet count).
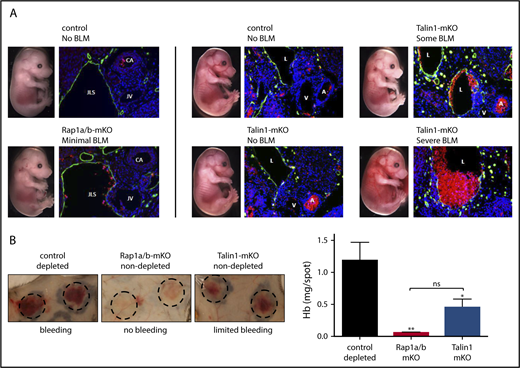
Platelet RAP1 signaling minimally contributes to the maintenance of vascular integrity during development and inflammation. (A) Contribution to vascular integrity during development was determined by assessing blood lymphatic mixing (BLM) in embryos at ∼E16.5 by macroscopic observation, using a Leica MZ16FA dissecting stereoscope, and immunofluorescence staining of embryo sections. Embryos were sectioned at the jugular lymph sac (JLS), and lymphatic endothelial cells (green) were stained with primary antibody overnight (polyclonal rabbit anti-mouse LYVE-1), followed by donkey anti-rabbit Alexa Fluor 488 and 4′,6-diamidino-2-phenylindole. Red blood cells were visualized by autofluorescence (red). Images were acquired on a Nikon E800 microscope with a Hamamatsu camera with MetaMorph software (Molecular Devices). (B) Contribution to vascular integrity at sites of inflammation was determined by rpA reaction in the skin of control (Pf4-Cre-) mice depleted of all circulating platelets (by intravenous injection of antibodies against GPIbα) compared with nondepleted Rap1a/b-mKO or nondepleted Talin1-mKO mice. Representative images of rpA sites are shown (dashed circles) (left panels). Hemorrhage at sites of inflammation was quantified by measuring hemoglobin (Hb) levels in skin lesions 4 hours after rpA challenge (n = 5-7) (right panel). *P < .05, **P < .01. ns, not significant.
Platelet RAP1 signaling minimally contributes to the maintenance of vascular integrity during development and inflammation. (A) Contribution to vascular integrity during development was determined by assessing blood lymphatic mixing (BLM) in embryos at ∼E16.5 by macroscopic observation, using a Leica MZ16FA dissecting stereoscope, and immunofluorescence staining of embryo sections. Embryos were sectioned at the jugular lymph sac (JLS), and lymphatic endothelial cells (green) were stained with primary antibody overnight (polyclonal rabbit anti-mouse LYVE-1), followed by donkey anti-rabbit Alexa Fluor 488 and 4′,6-diamidino-2-phenylindole. Red blood cells were visualized by autofluorescence (red). Images were acquired on a Nikon E800 microscope with a Hamamatsu camera with MetaMorph software (Molecular Devices). (B) Contribution to vascular integrity at sites of inflammation was determined by rpA reaction in the skin of control (Pf4-Cre-) mice depleted of all circulating platelets (by intravenous injection of antibodies against GPIbα) compared with nondepleted Rap1a/b-mKO or nondepleted Talin1-mKO mice. Representative images of rpA sites are shown (dashed circles) (left panels). Hemorrhage at sites of inflammation was quantified by measuring hemoglobin (Hb) levels in skin lesions 4 hours after rpA challenge (n = 5-7) (right panel). *P < .05, **P < .01. ns, not significant.
Discussion
Integrin inside-out activation is critical for platelet adhesion and hemostatic plug formation at sites of vascular injury. The dogma in the field is that integrin activation requires the assembly of the integrin-activation complex, including RAP1, TALIN1, and KINDLIN3, at the cytoplasmic tail of the integrin β subunit.48 However, although platelet integrin activation was completely abolished in mice deficient in Talin131,41 or Kindlin3,49 studies in germline-knockout mice identified a significant, but not crucial, role for RAP1B in this process.24,25 This led to the belief that there is a RAP1-independent mechanism that facilitates TALIN/KINDLIN3-mediated integrin activation in platelets.50 Alternative mechanisms for RAP1-independent TALIN recruitment in proximity of the integrin were suggested, including TALIN binding to acidic phospholipids.51 However, recent advances in proteomic profiling demonstrated that murine platelets also express ∼20 000 RAP1A molecules,52 a copy number that may be high enough to facilitate integrin activation in the absence of RAP1B. Our studies in mice lacking Rap1a and/or Rap1b in the megakaryocytic lineage provide the first definitive proof that RAP1A and RAP1B regulate the activation of αIIbβ3 and β1 integrins in platelets. Consistent with previous reports,24 integrin activation in platelets from Rap1b-mKO mice was reduced by >50%. However, we also observed a significant contribution of RAP1A to β1 and β3 integrin activation in stimulated platelets, which accounted for >70% of the integrin activation observed in platelets lacking RAP1B. The residual integrin activation observed in platelets from mice deficient in both RAP1 isoforms allowed for limited platelet aggregation, as measured by standard aggregometry. We and other investigators described similar results for mice expressing low levels of CalDAG-GEFI,34 mice expressing a loss-of-function (LOF) mutant of TALIN1,42 or mice expressing a LOF mutant of KINDLIN3.53 Our studies suggest that the minimal integrin activation observed in Rap1a/b-mKO platelets is not mediated by other RAP proteins, because RAP1-independent platelet aggregation did not correlate with RAP2 activation. Future studies will have to determine whether small GTPase-dependent or -independent mechanisms mediate limited integrin activation in the absence of RAP1 proteins. However, irrespective of the exact mechanism, the pathophysiological significance of this RAP1-independent pathway of integrin activation in arterial thrombosis and classical hemostasis is limited, considering that thrombus formation under physiological flow conditions was abolished in Rap1a/b-mKO mice. Our data indicate that expression of ≥1 Rap1 allele is necessary to allow for the minimal amount of integrin activation that supports the formation of 3-dimensional hemostatic plugs capable of ensuring hemostasis. Although expression of a single Rap1b allele in mice was sufficient to facilitate hemostasis at sites of small (laser) and large (tail-clip) injuries, expression of a single Rap1a allele could only support hemostatic plug formation after small injuries. At the moment, it is not clear how RAP1A and RAP1B mediate platelet integrin activation. It was suggested that the RAP1 effector RIAM was necessary to connect RAP1 and TALIN, thereby enabling the recruitment of TALIN to the plasma membrane where active RAP1 is localized.40 However, this hypothesis was disproved, because RIAM was found dispensable for β1 and β3 integrin activation, adhesion, and aggregation of murine platelets.54,55 Recent in vitro studies suggest that active RAP1 can directly bind TALIN,56 without an intermediate effector, but whether this is also true in platelets needs to be confirmed.
Our studies also provide indirect evidence for nonoverlapping roles for RAP1A, RAP1B, and RAP2 in platelet activation (supplemental Figure 2). Although both isoforms exhibit redundant function in integrin activation and TxA2 formation, RAP1B primarily facilitates cross talk with RAC1 and solely regulates α-granule secretion in platelets stimulated via the collagen receptor, GPVI. Interestingly, TxA2 formation was not completely abolished in Rap1a/b-mKO platelets, as it was in platelets lacking CalDAG-GEFI and P2Y12, pathways that control the activity state of RAP1 and RAP2.23 Consistent with a role for RAP2 in this process, proteomics profiling6,52 revealed that platelets express several RAP2-specific effectors,57,58 including MINK1. Platelets from mice deficient in MINK1 are defective in ERK and p38 MAPK signaling,59 pathways that contribute to TxA2 generation.60 Thus, although RAP2 does not seem to be involved in the regulation of integrin activation, it is a likely player in the regulation of MAPK-dependent TxA2 generation. Alternatively, TxA2 production may depend, in part, on a RAP-independent signaling pathway. Rap1a/b-mKO platelets, but not Rap1a-mKO and Rap1b-mKO platelets, were also defective in cellular responses depending on integrin outside-in signaling. Our data on Rap1b-mKO platelets is not consistent with previous work showing defects in outside-in signaling in platelets from Rap1b−/− mice.25 Aside from methodological differences, we do not have an explanation for the conflicting phenotypes. It is also important to remember that integrin inside-out activation has to precede outside-in signaling in spreading and clot-contraction assays. Rap1a-mKO and Rap1b-mKO platelets, but not Rap1a/b-mKO platelets, showed robust inside-out activation of αIIbβ3 integrin. Thus, the defect in spreading and clot contraction observed in Rap1a/b-mKO platelets may primarily be a reflection of impaired integrin activation in these cells. Consistent with this conclusion, Rap1b-mKO platelets exhibited a defect in RAC1 activation at an early time point after agonist stimulation, whereas no defect was seen later in the activation process. RAC1 activation was markedly diminished in Rap1a/b-mKO platelets throughout the activation process. These findings could be explained by normal outside-in signaling in RAP1 mutant platelets. More in-depth studies will be required to clarify this point.
Although well-established in platelets, surprisingly little is known about the contribution of RAP GTPase signaling to megakaryocyte development and platelet production. It is known that ERK is activated in response to megakaryocyte stimulation with thrombopoietin, a key cytokine regulator of megakaryocyte development,61,62 and that RAP and RAS GTPases mediate ERK activation in megakaryocyte cell lines via engagement of their downstream effectors B-RAF and RAF-1.63,64 Although mice deficient in RAF-1 did not show defects in megakaryocyte biology,65 deficiency in B-RAF led to impaired hematopoietic progenitor cell development and altered megakaryopoiesis.66 Furthermore, the cross talk between RAP1 and RAC1 may also affect platelet production, because RAC1 is an important regulator of proplatelet formation in mature megakaryocytes.67 However, deletion of RAC1 in mice did not affect the peripheral platelet count, unless another RHO GTPase, CDC42, was also deleted. Previous preclinical and clinical data did not suggest a role for RAP in megakaryocyte biology, because deficiency in CalDAG-GEFI or RAP1B did not affect the peripheral platelet count in mice, dogs, or humans.17,20,24,68 Consistent with these findings, a normal platelet count was observed in mice with megakaryocyte-specific deficiency in Rap1a or Rap1b. However, Rap1a/b-mKO mice exhibited a significant macrothrombocytopenia and a marked defect in proplatelet formation in vitro. Thus, we here provide the first definitive proof that RAP is a critical player during platelet production. Studies in progress will address whether RAP1A and RAP1B have redundant or unique functions during megakaryocyte development and proplatelet formation. Given its crucial role in platelet function, it is surprising that CalDAG-GEFI is dispensable in megakaryocytes. However, although RAP1 activation in platelets depends on a highly sensitive and rapidly activated GEF like CalDAG-GEFI, megakaryocytes do not depend on the same kinetics of cellular activation. Future studies should investigate the role of other RAP-GEFs, such as PDZ-GEF and EPAC, as regulators of RAP signaling in megakaryocytes.
In addition to their role in thrombus formation at sites of injury, platelets contribute to hemostasis by ensuring vascular integrity where the endothelial barrier has been breached without mechanical injury, for instance during development or at sites of inflammation in adulthood.69 In both cases, it is not clear how platelets affect vascular integrity. However, previous studies identified mechanistic similarities, because mice with defects in platelet ITAM signaling show BLM during embryonic development and marked hemorrhage at sites of inflammation.36,47,70-72 No bleeding was observed in mice with defects in receptors and signaling molecules important for classical hemostatic plug formation, including PAR and P2Y receptors, GPIb-V-IX, αIIbβ3, and CalDAG-GEFI.47 Interestingly, degranulated platelets failed to prevent tumor-associated inflammatory bleeding,73 suggesting that granule release, but not integrin-mediated adhesion, is critical for vascular integrity in inflammation and during development, a conclusion that was recently challenged by studies in mice genetically engineered to be defective in granule secretion.74 The studies reported here shift the focus back to integrins, because we observed significant BLM and inflammatory bleeding in Talin1-mKO mice; however, vascular integrity was intact in Rap1a/b-mKO mice. Considering this partial TALIN1 dependency and recent studies demonstrating that single platelets, not platelet aggregates, plug holes in the inflamed vascular wall,72 we propose that integrins other than αIIbβ3 or other TALIN1-regulated adhesion receptors contribute to platelet adhesion in this form of hemostasis. However, the very limited ability of Rap1a/b-mKO platelets to activate integrin receptors seems sufficient to prevent hemorrhage during embryonic development and inflammation. Alternatively, it is possible that TALIN1 has RAP1-independent functions that are critical for platelet-mediated vascular integrity.
In humans, LOF mutations in CalDAG-GEFI20,21,75-77 or P2Y12,78 the main RAP-stimulating pathways, lead to a marked defect in hemostasis. Interestingly, no patients with mutations in 1 of the RAP1 isoforms have been identified. Studies in RAP1-mutant mice24 suggested that LOF in Rap1b may cause embryonic lethality in humans, whereas mutations in Rap1a would be without significant impact on platelet function and hemostasis. However, it is important that RAP1A is much more abundant in human than in mouse platelets, with the expression ratio for RAP1B/ RAP1A being 2:1 in humans6 and 10:1 in mice.52 Our findings of functional redundancy between the 2 RAP isoforms in the inside-out activation of platelet integrins provide an alternative, more plausible explanation for the lack of LOF mutations in Rap1a or Rap1b in humans. We propose that both isoforms would have to be defective simultaneously for classical hemostasis to be impaired. The same conclusions can be drawn for the role of RAP1 in platelet production and the effect of RAP1 mutations on the peripheral platelet count. Lastly, our findings also have important implications for the development of novel antiplatelet therapies, because targeting of the pathways regulating RAP1 activity or of individual RAP1 isoforms would be expected to provide significant protection from thrombosis without jeopardizing the platelet count or hemostasis.
In conclusion, our studies demonstrate that RAP1 GTPases have redundant and isoform-specific functions in platelets and megakaryocytes. Mice deficient in both Rap1a and Rap1b exhibit significant macrothrombocytopenia due to impaired proplatelet formation, strongly impaired integrin activation in platelets, and marked defects in hemostasis after mechanical injury. In contrast, platelet RAP1 signaling is dispensable for the maintenance of vascular integrity during development and at sites of inflammation in mice.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Brian Cooley (Animal Surgery Core Laboratory, University of North Carolina at Chapel Hill) for assistance with the FeCl3 thrombosis experiment, Brian Petrich (Emory University) for providing floxed Talin1 mice and valuable expertise, Alexi Morozov (Virginia Tech) for generating floxed Rap1a/Rap1b mice, Lawrence Quilliam (Indiana University) for providing floxed Rap1a/Rap1b mice, and Keith Burridge for providing valuable reagents.
This work was supported by National Institutes of Health, National Heart, Lung, and Blood Institute grants R01 HL121650 and R01 HL130404 (W.B.), R01 HL133668 (W.B. and K.M.H.), and T32 HL007149 (R.H.L.); British Heart Foundation grant RG/15/2/31224 (J.M.G.); and the Italian Ministry of Education, University and Research’s Rita Levi Montalcini Programme (L.S.).
Authorship
Contribution: L.S. designed research, performed research, analyzed data, and wrote manuscript; R.H.L. performed experiments, analyzed data, and wrote manuscript; D.S.P., D.G., Y.B., R.P., and D.O.K. performed experiments; E.C.O. and C.I.J. performed experiments and analyzed data; K.O.P. performed experiments and provided technical assistance; K.M.C. analyzed data; K.M.H. contributed vital reagents; J.M.G. contributed vital reagents and analyzed data; and W.B. designed research and wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Wolfgang Bergmeier, University of North Carolina, 120 Mason Farm Rd, Campus Box 7260, Chapel Hill, NC 27599; e-mail: bergmeie@email.unc.edu; and Lucia Stefanini, Sapienza University of Rome, Viale del Policlinico, 155, 00161 Rome, Italy; e-mail: lucia.stefanini@uniroma1.it

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal