

Abstract
The complex pathophysiology in β-thalassemia can translate to multiple morbidities that affect every organ system. Improved survival due to advances in management means that patients are exposed to the harmful effects of ineffective erythropoiesis, anemia, and iron overload for a longer duration, and we started seeing new or more frequent complications in adult compared with younger patients. In this article, we highlight particular aspects of managing adult patients with β-thalassemia, using our own experience in treating such patients. We cover both transfusion-dependent and nontransfusion-dependent forms of the disease and tackle specific morbidities of highest interest.
Introduction
The β-thalassemias, a group of inherited hemoglobin disorders, continue to be a concern for health care systems owing to the high burden of disease and its management.1-3 The severity of ineffective erythropoiesis and subsequent anemia depends on several genetic and environmental factors and the disease phenotype was historically labeled as major, intermediate, or minor accordingly.1 However, in more recent years, we started categorizing patients according to their transfusion requirement, to optimize practical management considerations, although β-thalassemia should always be considered a spectrum of severities with patients able to move from one to another as with management or natural progression of disease.4 Transfusion-dependent β-thalassemia (TDT) patients commonly present to our clinics in early childhood with severe anemia that requires lifelong regular transfusion therapy for survival. Nontransfusion-dependent β-thalassemia (NTDT) patients usually present later in childhood or even in adulthood with mild/moderate anemia that only requires occasional or short-course regular transfusions in certain clinical settings. Recent management guidelines have also taken this direction of classification into TDT and NTDT in their recommendations.1,5-7
Over the last few decades, there has been a considerable advance in understanding the disease process of β-thalassemia, and key milestones in optimizing management with transfusion or iron chelation have been achieved. Such advances in supportive management led to a significant improvement in survival in this once fatal disease.8,9 For example, mortality rates in western cohorts have declined from 12.7 to 1.65 deaths per 1000 patient-years between the periods 1980 to 1999 and 1999 to 2013 with the leading cause of death moving from iron overload and bone marrow transplant complications to infections and hepatitis C virus complications.10,11 However, such advances could not completely abolish the underlying pathophysiology, which meant that several morbidities continued to manifest at higher incidence with advancing age and chronic exposure to risk factors. Moreover, increased awareness of the disease process prompted clinicians to apply closer and more regular monitoring, which usually leads to higher detection of preclinical and clinical complications especially in adulthood. With this background, we herein share our experience in managing complications in adults with β-thalassemia, especially in their mid-30s and beyond. We limited our coverage to select complications that we most commonly see in our clinics or those persistently reported at higher incidence with advancing age in the literature. It should be noted, however, that such morbidities can still manifest in younger patients, especially in those with severe forms of the disease. Lastly, as clinical trials in this context are limited, we mostly relied on observational data from our own clinics or our expert opinion stemming from direct patient experience.
General management considerations in TDT
In patients with TDT, the culprit of disease process is secondary iron overload from regular transfusion therapy, which can lead to organ damage and failure especially in the heart, liver, and endocrine glands.12 With advances in magnetic resonance imaging (MRI) that allowed noninvasive estimation of iron levels in key target organs,13,14 we realized that significant iron accumulation in these organs can start from early childhood15-18 and continues to accumulate over time if not optimally treated, leading to the emergence of clinical morbidities.19 It is hard to assign a specific age of incidence for the various potential complications, as it relies on the specific transfusion and iron chelation practice and patient response. In suboptimally treated patients, we commonly experience an early onset of endocrine disorders in childhood, adolescence, or early adulthood (growth failure, hypogonadism) with an increasing risk as patients age, in view of cumulative exposure as well as the underlying increased risk seen in the general aging population (eg, hypothyroidism, hypoparathyroidism, diabetes, osteoporosis). Heart failure and arrythmias can be detected in early adulthood, again with an increasing risk as patients naturally age, whereas we more commonly tend to see arrythmias in older adults. Early signs of hepatic enzymes that increase secondary to iron overload can be detected at any age, whereas overt hepatic disease such as fibrosis, cirrhosis, or hepatocellular carcinoma are more time-dependent and we more commonly see them in older adults. Management of arrythmias and hepatic disease in adult patients is covered separately in “Specific morbidities.” In a nutshell, early optimization of transfusion and iron-chelation therapy from childhood toward adulthood is essential to ensuring adequate red blood cell supply while still preventing cumulative iron toxicity and subsequent morbidities, especially in later adulthood. More importantly, close and regular monitoring for clinical complications can ensure early detection, specialist consultation, and intervention.6 In Figure 1, we highlight key clinical and psychosocial concerns across age groups for patients with TDT. In Figure 2, we highlight standard monitoring recommendations to ensure early detection of abnormalities, although these could be adapted for specific patient populations with varying requirement. Monitoring recommendations for specific morbidities are further discussed in “Specific morbidities.”
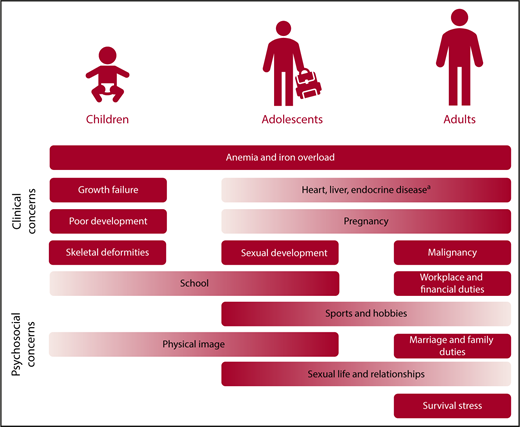
Management priorities across age groups in TDT. The listing reflects potential incidence of morbidities in respective age groups, but is not exclusive, and individual patients may have different needs. aIncludes diabetes mellitus, hypothyroidism, hypoparathyroidism, osteoporosis, hypogonadism.
Management priorities across age groups in TDT. The listing reflects potential incidence of morbidities in respective age groups, but is not exclusive, and individual patients may have different needs. aIncludes diabetes mellitus, hypothyroidism, hypoparathyroidism, osteoporosis, hypogonadism.
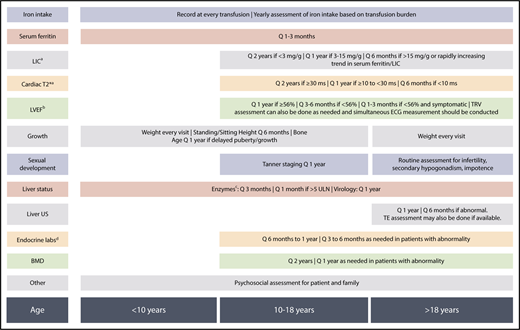
General monitoring recommendations across age groups in TDT.aBy MRI R2 for liver or MRI T2* for liver and heart with appropriate calibration. LIC and cardiac T2* may be assessed at earlier age (from 6 years) if feasible especially in patients who are on high iron intake. bAs assessed by experienced echocardiographer or cardiac MRI. cAlanine aminotransferase, aspartate aminotransferase, total and direct bilirubin. dThyroid-stimulating hormone; calcium, phosphate, vitamin D, and parathyroid hormone (as indicated); luteinizing hormone, follicle-stimulating hormone, testosterone, estradiol, gonadotropin-releasing hormone (as indicated in cases of abnormal sexual development); fasting blood sugar, oral glucose tolerance test (as indicated). BMD, bone mineral density; ECG, electrocardiogram; LIC, liver iron concentration; LVEF, left-ventricular ejection fraction; Q, every; TE, transient elastography; TRV, tricuspid-regurgitant jet velocity; ULN, upper limit of normal; US, ultrasound.
General monitoring recommendations across age groups in TDT.aBy MRI R2 for liver or MRI T2* for liver and heart with appropriate calibration. LIC and cardiac T2* may be assessed at earlier age (from 6 years) if feasible especially in patients who are on high iron intake. bAs assessed by experienced echocardiographer or cardiac MRI. cAlanine aminotransferase, aspartate aminotransferase, total and direct bilirubin. dThyroid-stimulating hormone; calcium, phosphate, vitamin D, and parathyroid hormone (as indicated); luteinizing hormone, follicle-stimulating hormone, testosterone, estradiol, gonadotropin-releasing hormone (as indicated in cases of abnormal sexual development); fasting blood sugar, oral glucose tolerance test (as indicated). BMD, bone mineral density; ECG, electrocardiogram; LIC, liver iron concentration; LVEF, left-ventricular ejection fraction; Q, every; TE, transient elastography; TRV, tricuspid-regurgitant jet velocity; ULN, upper limit of normal; US, ultrasound.
Although the 3 available iron chelators deferoxamine, deferiprone, and deferasirox have served patients well and have a large body of evidence for efficacy in chelating iron from target organs,6,20 we still see a considerable number of patients with high liver and cardiac iron concentration globally,21 especially in older adults who were suboptimally treated in childhood due to poor adherence to subcutaneous deferoxamine and imprecise iron measurement tools. Thus, as much as the onus is to prevent iron accumulation by optimal treatment from childhood, adult patients would require adequate chelation to decrease high iron levels to safety thresholds. All 3 iron chelators have established efficacy in significantly reducing iron levels from the liver and heart as evident from trials applying MRI technology.6,22-29 The question of which iron chelator is more effective or suitable as first line is futile, as management of patients should be individualized, and different iron chelators may be suitable for different iron overload profiles. Optimal dosing, side-effect monitoring, and adherence are essential for any used iron chelator. In patients with established heart failure, we rely on continuous 24-hour deferoxamine infusion, which was shown to improve cardiac function in earlier studies of TDT patients.30 Similar observations of cardiac function improvement and reversal of heart failure were noted in trials of deferiprone monotherapy and the combination of deferiprone and deferoxamine.31-33 The details of management of cardiac complications can be found in the American Heart Association consensus statement on cardiac disease in thalassemia.34 Deferasirox is currently the only chelator to demonstrate stabilization or improvement in hepatic fibrosis.35 Nonclinical trial data have also demonstrated the ability of deferasirox or combination therapy in stabilization or reversal of endocrine and bone disease.36,37
General management considerations in NTDT
Anemia in NTDT can be progressive with age and is directly and independently associated with increased morbidity.38-40 Ineffective erythropoiesis also leads to a state of primary iron overload in the absence of transfusion therapy, driven by hepcidin dysregulation.41,42 Iron overload is cumulative over time and as patients advance in age,38,43,44 with iron overload indices reaching clinically critical thresholds and associated with several morbidities in observational studies including hepatic fibrosis, thrombosis, pulmonary hypertension, endocrinopathies, osteoporosis, and cerebrovascular disease, with notable absence of cardiac iron loading.45,46 Ineffective erythropoiesis can also lead to progressive bone marrow expansion and bone changes or mineral density reduction as well as hypercoagulability due to hemolyzed prothrombotic red cell production, the latter leading to vascular events.5,47-50 Thus, without intervention, NTDT patients experience increased morbidity as they advance in age with a notable incidence beyond the age of 35 years,38,40 with some complications that are less commonly seen in patients with TDT (Figure 3). It should also be noted that quality of life is directly related to age and multiplicity of morbidity in NTDT; hence, attention and appropriate psychological care should be administered.51 Management of select morbidities that we commonly encounter in adult patients at our clinics will be featured in “Specific morbidities.”
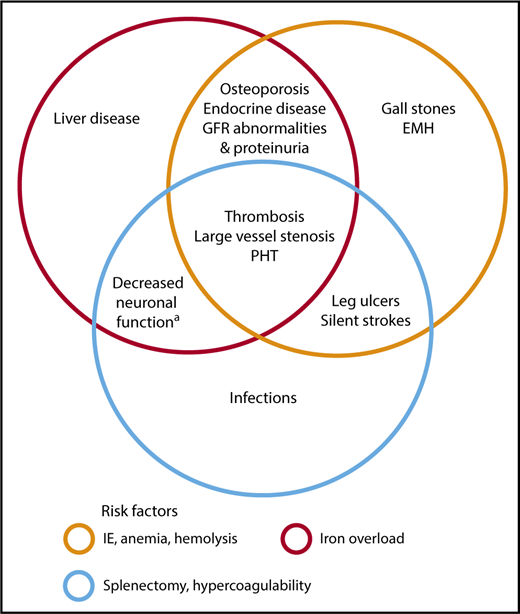
Morbidities and risk factors in patients with NTDT. Risk factors and pathophysiologic mechanisms as Venn diagrams within which notable morbidities associated with them are included. Some morbidities are attributed to >1 risk factor. These associations are mostly based on data from observational studies. aAs evident on fluorodeoxyglucose positron emission tomography–computed tomography (PET-CT). EMH, extramedullary hematopoietic pseudotumors; GFR, glomerular filtration rate; IE, ineffective erythropoiesis, PHT, pulmonary hypertension.
Morbidities and risk factors in patients with NTDT. Risk factors and pathophysiologic mechanisms as Venn diagrams within which notable morbidities associated with them are included. Some morbidities are attributed to >1 risk factor. These associations are mostly based on data from observational studies. aAs evident on fluorodeoxyglucose positron emission tomography–computed tomography (PET-CT). EMH, extramedullary hematopoietic pseudotumors; GFR, glomerular filtration rate; IE, ineffective erythropoiesis, PHT, pulmonary hypertension.
In patients with NTDT, no formal clinical trials evaluated the role of regular transfusion therapy as much as occasional transfusions are used in cases of pregnancy or surgery or during infections.7 We commonly restrict our use of regular transfusion therapy to specific clinical settings, such as in cases of growth failure or poor sexual development in childhood and adolescence. For adults, consideration of regular transfusion courses is also suggested for prevention or management of certain morbidities (thrombotic events, pulmonary hypertension, leg ulcers, extramedullary hematopoietic pseudotumors) in high-risk patients, backed up by data on transfusion benefit from observational studies of our own patients.7,40 The need for transfusion therapy already indicates a more severe phenotype in NTDT that may warrant the need for continuous transfusions if the risk/benefit ratio is favorable.7 Careful attention should be paid to the potential risks of iron overload and alloimmunization (especially in splenectomized, pregnant, or previously never-transfused patients).7 We almost stopped relying on splenectomy at all owing to the large body of evidence on an increase of a variety of morbidities as well as infections in splenectomized patients.7,40 We mainly restrict it to patients with symptomatic splenomegaly or hypersplenism. Iron chelation became standard of care for all patients over 10 years of age with iron overload.7 We commonly check all patients older than 10 years for iron overload with serum ferritin or MRI depending on cost and availability. Patients with iron overload (serum ferritin > 800 µg/L or liver iron concentration > 5 mg Fe/g dry weight)43,52 are started on iron chelation therapy with deferasirox.53-55 We follow dosing and iron assessment schedules from management guidelines.7
Specific morbidities
In this section, we will feature clinical cases to illustrate the course and management of specific morbidities in patients with TDT or NTDT. These have been selected based on our experience of encountering them most commonly in adults with β-thalassemia, although they may still manifest earlier especially in severe or poorly managed patients. Other clinical complications that may manifest earlier in the natural course of β-thalassemia and that also may increase in incidence with advancing age (eg, heart failure, endocrine and bone disease, leg ulcers, extramedullary pseudotumors, gallstones, infections) have been featured elsewhere, and management guidelines for those are widely available.6,7,56 Lastly, we should also highlight that with advanced age comes an added risk of complications such as cancer, similar to the general population. One epidemiologic study found that the incidence of cancer (3.96 per 1000 person-years) in thalassemia patients was 52% higher than the general population, especially for hematological and abdominal malignancies.57 Iron overload and hepatitis C virus infection increase the risk of hepatocellular carcinoma in thalassemia, whereas oxidative stress in the bone marrow is suggested to increase the risk of hematologic malignancies.58,59
Case 1: arrythmia
A 42-year-old man with TDT was admitted for evaluation of cardiac arrythmia. He was receiving 2 to 3 units of packed red blood cells every 21 days with a mean pretransfusional hemoglobin of 10.5 g/dL. His comorbidities included glucose intolerance, hypogonadotropic hypogonadism, and subclinical hypothyroidism. He started iron chelation therapy at 4 years of age with subcutaneous deferoxamine. Five years prior to the current presentation, his cardiac T2* MRI showed a moderate myocardial iron overload (14.4 ms), and deferiprone (75 mg/kg per day) was added to his iron chelation regimen in combination with deferoxamine (30 mg/kg per day). His cardiac T2* showed improvement to 24.5 ms 3 years after, and continues to be over 25 ms according to the latest imaging. He stared experiencing several episodes of arrythmia over the last 2 years (atrial tachycardia with 2:1 atrioventricular block/atrial flutter), which required frequent hospitalizations including the present one. Routine echocardiography has never shown any abnormalities or impairment of left ventricular function except for a mild/moderate atrial dilatation. Subsequently, the patient was sent for catheter ablation. His last 24-hour electrocardiogram (ECG) showed normal sinus rhythm with atrioventricular block (grade 1). He was discharged on bisoprolol 1.25 mg twice daily and amiodarone 200 mg once daily.
Arrythmia
With advances in cardiac MRI imaging and the availability of oral iron chelators with improved adherence and efficacy in cardiac iron removal, the incidence of heart failure and its associated fatality in TDT patients continue to decline.6,10 However, this delay or prevention in heart failure allowed other cardiac manifestations to become more apparent in older adults with thalassemia, such as arrythmias (especially atrial fibrillation with a prevalence of ∼40% in patients over 40 years), cardiac function changes due to restriction, fibrosis, and arterial changes due to loss of vascular compliance.6 Symptomatic arrhythmias in thalassemia patients pose a significant clinical risk and are associated with significant myocardial iron overload. They can also occur in patients with otherwise normal iron levels, this being attributed to fibrosis from past iron deposition that was cleared.6 Patients should be assessed by history taking for symptoms on every visit, and ECGs should be done annually for all patients, biannually in asymptomatic patients with abnormality, and monthly or more frequently in symptomatic patients. Assessment can follow the course of echocardiographic or cardiac T2* measurement and should not be ignored. The emerging role of MRI measurement of cardiac fibrosis can also be considered.
In the presence of known or suspected severe cardiac iron overload, we intensify iron chelation therapy when these symptomatic arrythmias manifest, even if cardiac MRI assessment of myocardial iron status is not feasible, especially as a matter of urgency if the symptoms include syncope or presyncope. Arrhythmias in TDT patients can often be controlled or eliminated by aggressive iron chelation, with IV deferoxamine or high-dose deferiprone monotherapy or a combination.60 However, this may require some time, and short-term antiarrhythmic therapy is needed in consultation with a cardiologist in view of the constantly changing practice in rhythm control, and the different approaches needed for different arrythmias, although with low evidence, amiodarone has been used in the limited setting of inpatient and acute care because of its broad spectrum of action and modest compromise of cardiac function.34 It should be noted, however, that amiodarone therapy is associated with side effects especially relevant for patients with thalassemia, particularly for liver and thyroid function that would require close monitoring. Beta-blockers are generally well tolerated, if titrated slowly, and can be useful in controlling ectopic rhythms.34 Catheter ablation may be considered in patients with chronic symptomatic arrhythmias, but patients should be referred to a cardiac electrophysiology specialist before ablation, given that fibrosis can be challenging to ablate.
It should be noted that arrythmias may not always be caused by iron overload in β-thalassemia, particularly in younger adults who present with supraventricular arrhythmia. In such cases, other causes such as alcohol or substance abuse or excess of thyroid hormones should be ruled out.
Case 2: liver disease
A 42-year-old man with TDT presented with right upper quadrant pain that has been persistent for 4 months. Initial imaging with contrast-enhanced computed tomography showed foci in his liver that were initially interpreted as extramedullary hematopoiesis. Liver function tests were within normal range as well as albumin level and prothrombin time. His ferritin level was 3100 µg/L; his liver iron concentration, also measured by MRI, was 17.3 mg Fe/g dry weight. A computed tomography–guided biopsy was recommended and showed the lesions to be compatible with multifocal hepatocellular carcinoma. Hepatitis B and C testing by polymerase chain reaction were negative. The patient received palliative treatment. He developed hepatorenal syndrome and rapidly succumbed to his illness.
Liver disease
Liver disease is becoming a leading cause of mortality in TDT, especially owing to the decline in mortality from heart failure associated with advances in cardiac iron monitoring and effective chelation therapy.61 Liver disease is also responsible for ∼10% of the causes of death in NTDT patients.62 The liver is the primary site of storage for excess iron; hence, in the absence of effective iron chelation therapy, liver iron concentration can reach clinically critical levels in both TDT and NTDT patients, although at slower rates in NTDT. Primary iron overload from increased intestinal iron absorption in NTDT leads to preferential portal and hepatocyte iron loading whereas hepatic loading in TDT patients also involved the reticuloendothelial system.6,7 Irrespectively, chronic hepatic iron deposition promotes liver fibrogenesis and cirrhosis in both patient populations.43,52,63-66 A longer duration of hepatic iron exposure is associated with a higher risk of significant fibrosis and cirrhosis, which is why overt morbidity primarily manifests in older adults.67,68 In regularly transfused patients, hepatic disease may also be caused or exacerbated by hepatitis C (and less commonly B) virus infection. Although the incidence of viral hepatitis substantially declined after the introduction of regular donor-screening programs in the 1990s and further with the introduction of RNA–polymerase chain reaction testing, adults who may have acquired it earlier continue to have a high prevalence. Hepatitis C virus infection and iron overload are independent risk factors for hepatic fibrosis and cirrhosis, although their coexistence is synergistic in injury.66
Both hepatic iron overload and chronic hepatitis C virus (and less so hepatitis B virus) infection can lead to hepatocellular carcinoma. The carcinogenicity of iron is related to its induction of oxidative damage, which results in genotoxicity, and to immunologic dysregulation, which attenuates cancer immune surveillance. Chronic hepatitis C infection leads to necroinflammation, which can prompt progression to cancer.69 Several cases of hepatocellular carcinoma in patients with TDT and NTDT from our clinics or those of colleagues have been described in the literature. Most cases occurred in older adults (>45 years) who had cirrhosis and viral hepatitis infection, although malignancy occurred in some patients in the absence of viral hepatitis or cirrhosis while having considerably high iron overload levels.69-74 Additional factors such as obesity or alcohol consumption may also increase steatosis and oxidative stress, which accelerate liver iron uptake and increase risk of liver fibrosis, cirrhosis, and cancer in β-thalassemia patients.75
Our approach for the diagnosis and management of liver disease in β-thalassemia is summarized in Figure 4. The hallmark is close monitoring not only for iron overload levels or hepatitis infection status but also for hepatic injury. Optimal management of iron overload is key to preventing liver toxicity. All 3 available iron chelators have data on efficacy in hepatic iron reduction on MRI, although deferasirox has the largest body of evidence in severely iron-loaded patients (liver iron concentration > 15 mg Fe/g dry weight)25-27 and has demonstrated ability to reduce necroinflammation and fibrosis in a significant proportion of patients on 3 years’ therapy.35 Management of viral hepatitis should be done in consultation with an experienced hepatologist, especially in pregnant women, immunosuppressed patients, and those with cirrhosis.6,7 Earlier studies of antiviral therapy in β-thalassemia with peg-interferon and ribavirin showed sustained virological responses in 25% to 64% of patients.76 However, ribavirin is known to be associated with worsening anemia and increased transfusion demands, which is a major concern in β-thalassemia patients. Moreover, most clinicians today opt to go for interferon-free regimens. The remarkable revolution in hepatitis C management with direct-acting antiviral drugs offers a new opportunity for β-thalassemia to receive once-daily treatments with no need to check for genetic polymorphisms, and with treatment schedules typically lasting 8 to 12 weeks (eg, glecaprevir-pibrentasvir, ledipasvir-sofosbuvir, or sofosbuvir-velpatasvir for genotype 1). In observational studies and small trials, sustained virological responses at 12 weeks were reported in up to 90% to 100% irrespective of cirrhosis status.77-80 In a recent review, data compiled from 420 thalassemia patients across different studies, with genotypes 1a, 1b, 2, 3, and 4, showed a success rate of at least 93% with direct-acting antiviral drugs.81 Thus, hepatitis C virus infection in β-thalassemia today is definitely treatable, and much more conveniently than before.
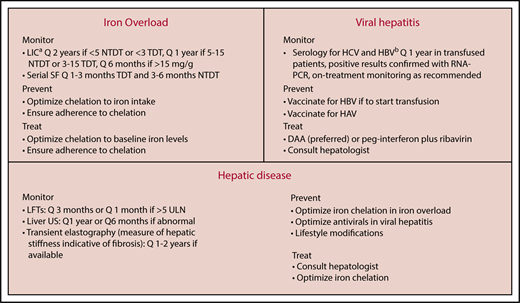
Approach to monitoring and management of liver disease in adult β-thalassemia patients.aBy MRI R2 or T2*, the latter requiring appropriate calibration and preferred if cardiac iron assessment is required at the same time. MRI monitoring can start at the age of 10 years in TDT (or earlier if feasible and deemed necessary) and NTDT. bIf not anti-HBsAg+. DAA, direct-acting antiviral drug; HAV, hepatitis A virus; HBV, hepatitis B virus; HCV, hepatitis C virus; LFT, liver function test; LIC, liver iron concentration; RNA-PCR, RNA polymerase chain reaction; SF, serum ferritin.
Approach to monitoring and management of liver disease in adult β-thalassemia patients.aBy MRI R2 or T2*, the latter requiring appropriate calibration and preferred if cardiac iron assessment is required at the same time. MRI monitoring can start at the age of 10 years in TDT (or earlier if feasible and deemed necessary) and NTDT. bIf not anti-HBsAg+. DAA, direct-acting antiviral drug; HAV, hepatitis A virus; HBV, hepatitis B virus; HCV, hepatitis C virus; LFT, liver function test; LIC, liver iron concentration; RNA-PCR, RNA polymerase chain reaction; SF, serum ferritin.
Once manifested, hepatocellular carcinoma would be managed according to standard of care and stage of the disease using chemotherapy, targeted or immune therapy, radiofrequency ablation, transplant, or palliative care. Effective hepatitis C virus clearance in patients with hepatocellular carcinoma improves survival and recurrence rates, but the role of iron chelation in this setting has not been fully evaluated.82
Case 3: thrombotic disease
A 38-year-old splenectomized man with NTDT presented with severe right calf pain. The pain started 4 days prior to presentation, intensified gradually, and was associated with a sensation of hotness around the area. No history of trauma or fever was reported and no personal or family history of thrombosis was documented. A thrombophilia workup was also negative. The pain was unresponsive to analgesics. On physical examination, the patient had diffuse erythema in the right calf which was hot and tender to palpation. No edema was noted. Laboratory workup revealed a total hemoglobin level of 8.4 g/dL, a nucleated red blood cell count of 460 × 106/L, and a platelet count of 986 × 109/L. Duplex ultrasonography revealed deep thrombosis of the posterior tibial vein. The patient was started on a treatment dose of low-molecular weight heparin and was started on a course of regular transfusion therapy. Aspirin was also initiated.
Thrombotic disease
A hypercoagulable state has been identified in β-thalassemia patients.83,84 It is primarily attributed to abnormalities in platelets and pathological red blood cells, although several additional factors are believed to be involved, including endothelial dysfunction, coagulation system abnormalities, and presence of microparticles, ultimately leading to clinical thrombosis.50 The incidence of clinical thrombosis is fourfold higher in NTDT compared with TDT patients, is mostly venous, and is a leading cause of mortality.85,86 The frequency of thrombosis in NTDT is significantly higher in patients older than 35 years (28.2%) compared with patients 18 to 35 years (14.9%), or younger than 18 years (4.1%).40 Splenectomy, anemia (hemoglobin level < 9 g/dL), and iron overload (serum ferritin > 800 µg/L or liver iron concentration > 5 mg Fe/g dry weight) have all been identified as risk factors.40,43,52,85,87 Additionally, high nucleated red blood cell (≥300 × 106/L) and platelet counts (≥500 × 109/L) were shown to further substantiate the risk in splenectomized NTDT patients.86 Although the incidence of overt strokes is relatively low (∼5%),85,86 silent strokes have been described in up to 60% of splenectomized adults (mean age, 32 years) with NTDT, with a clear correlation with advancing age.88,89 The clinical significance of these white matter lesions or whether they require any intervention, however, is not yet clear.
We treat patients who develop thrombotic disease as per standard local or international guidelines for nonthalassemic patients. Patients who present with unprovoked, spontaneous thrombosis at unusual sites are also worked up for thrombophilia despite the established risk in thalassemia. For primary or secondary prevention, data on risk stratification or prophylaxis in thalassemia are lacking and the approach is mostly individualized. We commonly consider β-thalassemia patients as high risk in medical and surgical settings, especially patients with the aforementioned thalassemia-related (eg, NTDT, splenectomy, low hemoglobin, high platelet or nucleated red blood cell counts) and nonrelated (eg, older age, pregnancy, malignancy, immobility, previous history of thrombosis) risk factors. If prophylaxis is deemed necessary, we commonly use enoxaparin. Newer oral anticoagulants may also be considered, while also acknowledging lack of data in thalassemia, especially if long-term prophylaxis is needed. Blood transfusions may control the hypercoagulability in NTDT patients by improving ineffective erythropoiesis and decreasing the levels of pathological red blood cells with thrombogenic potential.90 In observational studies from our clinics, transfusion therapy has been associated with lower rates of thromboembolic events.40 It should be noted that a specific transfusion schedule or duration for such patients is not defined. In our practice, we often rely on less frequent regular transfusions to lower the risk of iron overload. These practices should be individualized and based on clinical patient response. The association between high platelet counts or platelet activation and thrombosis as well as a lower recurrence rate of thrombotic events in splenectomized NTDT patients who took aspirin after their first event, when compared with those who did not, suggests a potential role for aspirin in prevention.85,86 We commonly administer aspirin therapy in all splenectomized patients irrespective of platelet count.
Case 4: pulmonary hypertension
A 46-year-old splenectomized woman with NTDT presented with progressive dyspnea of 2 weeks’ duration. She had no prior history of cardiac disease. Her laboratory studies revealed a total hemoglobin level of 10.2 g/dL, a platelet count of 850 × 109/L, and a serum ferritin level of 1520 µg/L. On continuous-wave Doppler transthoracic echocardiography, she had a peak tricuspid-valve regurgitant jet velocity (TRV) of 3.5 m/s. She was referred to an interventional cardiologist and underwent a right heart cardiac catheterization that revealed a mean pulmonary arterial pressure of 45 mm Hg. She was started on anticoagulant and regular transfusion therapy. She was asked to present back for reevaluation with echocardiography in 6 months.
Pulmonary hypertension
Although the exact mechanisms implicated in the pathogenesis of pulmonary hypertension in β-thalassemia remain unclear, its association with anemia, hemolysis, and hypercoagulability as well as complex interactions of platelets, the coagulation system, erythrocytes, and endothelial cells along with inflammatory and vascular mediators are suggested.91-93 Pulmonary hypertension in β-thalassemia is pulmonary arterial hypertension, characterized by the presence of precapillary pulmonary hypertension in the absence of left-sided heart disease, lung disease, or chronic thromboembolism.94-96 However, the possibility of pulmonary hypertension occurring secondary to chronic thromboembolic disease cannot be fully excluded with the hypercoagulable state noted in β-thalassemia.97,98 In newer classification, it would belong to group 5 pulmonary hypertension associated with chronic hemolytic anemia with unclear/multifactorial mechanism.99
Historic studies relying primarily on echocardiographic criteria (TRV exceeding 2.5-2.8 m/s) to document pulmonary hypertension in β-thalassemia provide an overestimate of its prevalence.7,40 In a more recent large study of ∼1300 patients from Italy, the prevalence of pulmonary hypertension was considerably lower when more strict echocardiographic criteria and confirmatory right heart catheterization were used (5.7% for a TRV > 3.0 m/s, 3.6% for a TRV > 3.2 m/s, and 2.1% on right heart catheterization). Patients with NTDT had a fivefold increased prevalence compared with TDT (4.8% vs 1.1%).100 Right heart catheterization is needed to confirm the diagnosis. Even in studies relying on catheterization, a significant correlation was noted between prevalence and age, with an exponential rise noted after the age of 45 years.100 Splenectomy, a history of thrombosis, a platelet count ≥500 × 106/L, nucleated red blood cell counts ≥300 × 106/L, and iron overload (serum ferritin > 800 µg/L or liver iron concentration > 5 mg Fe/g dry weight) have all been reported as risk factors for pulmonary hypertension in NTDT patients.40,43,52,98,100
When manifest, pulmonary hypertension is associated with functional limitation and can lead to right-sided heart failure.97,100-106 Our approach for the diagnosis, prevention, and management of pulmonary hypertension in β-thalassemia is summarized in Figure 5. Although no clinical trials exist, a role for transfusion therapy in preventing the occurrence of pulmonary hypertension is suggested in observational studies, but the specific schedule and duration of use are not defined and should be individualized.40,98,107 Similar effects were also noted with hydroxyurea therapy40,98,108-111 and iron chelation therapy in observational studies.40,98 For management, sildenafil citrate showed promising results in small studies112-114 and trials by improving cardiopulmonary hemodynamics in patients with a TRV > 2.5 m/s.115 Its use, however, should be limited to patients with confirmed pulmonary hypertension by cardiac catheterization.
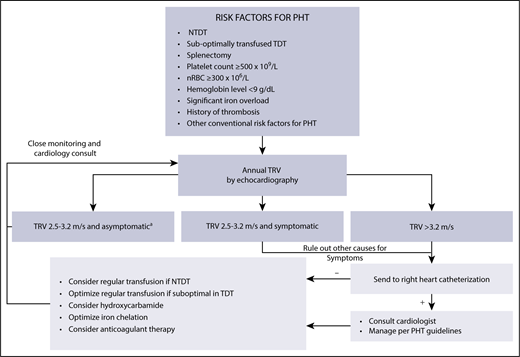
Approach to diagnosis, prevention, and management of pulmonary hypertension in adult patients with β-thalassemia.aPatients with TRV < 2.5 m/s may also be reassessed but at longer intervals (3-5 years). nRBC, nucleated red blood cell count; PHT, pulmonary hypertension.
Approach to diagnosis, prevention, and management of pulmonary hypertension in adult patients with β-thalassemia.aPatients with TRV < 2.5 m/s may also be reassessed but at longer intervals (3-5 years). nRBC, nucleated red blood cell count; PHT, pulmonary hypertension.
Future prospects
Several novel therapies are being developed for patients with β-thalassemia. Gene therapy to replace the defective β-globin gene has already shown promising results in reducing or eliminating the transfusion requirement in TDT patients,116 whereas genome editing to reinstate the capacity of fetal hemoglobin production is under way.117 JAK2 inhibitors have also shown some positive results in ameliorating splenomegaly in TDT, although with a modest effect on transfusion requirement.118 The ligand traps sotatercept and luspatercept have shown promising results in phase 2 studies with significant reduction in transfusion requirement for TDT patients and increase in hemoglobin level in NTDT patients; data from randomized clinical trials are awaited.119,120 Hepcidin agonists are also being evaluated for their ability to prevent iron overload and improve anemia in β-thalassemia patients.121 Reductions in transfusion requirement and subsequent prevention of iron overload in TDT and improvement in anemia with prevention of iron overload in NTDT will surely translate to reduced morbidity risk. Although we expect that such agents, if successful in clinical trials, can transform the disease picture for younger patients and become the mainstay of therapy, older patients who have been exposed to the damaging effects of anemia and iron overload for decades will continue to require conventional therapy for optimal management of their complications risk.
β-thalassemia is a disease of multiple risk factors and multiple morbidities, which logically implies the need for a multidisciplinary management team. This becomes particularly essential for older patients with comorbidities who require the attention of internists and specialists alongside their primary care. Transition from child into adult care facilities becomes more essential for older patients. That said, the ideal treatment strategy will always be an individualized one.
Acknowledgments
The authors thank Khaled Musallam (International Network of Hematology, London, United Kingdom) for constructive review of the manuscript.
Authorship
Contribution: A.T.T. and M.D.C. wrote the manuscript and gave final approval of the manuscript for submission.
Conflict-of-interest disclosure: A.T.T. received honoraria and research grants from Novartis and Celgene. M.D.C. received honoraria from Novartis and Celgene.
Correspondence: Ali T. Taher, Department of Internal Medicine, American University of Beirut Medical Center, P.O. Box 11-0236, Beirut 11072020 Lebanon; e-mail: ataher@aub.edu.lb.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal