

Abstract
With increasing survival, cumulative complications of sickle cell disease (SCD), which develop insidiously over time, are becoming more apparent and common in older patients, particularly those in their fifth decade and beyond. The older patient is also more likely to develop other age-related nonsickle conditions that interact and add to the disease morbidity. A common misconception is that any symptom in a SCD patient is attributable to their SCD and this may lead to delays in diagnosis and appropriate intervention. We recommend regular comprehensive reviews and monitoring for early signs of organ damage and a low threshold for the use of hydroxyurea and blood transfusions as preventative measures for end-organ disease. Treatable comorbidities and acute deterioration should be managed aggressively. Although the primary goal in management of the older adult with SCD is improving anemia and minimizing organ damage, the time has come for us to be more proactive in considering curative therapies previously offered to the younger patient. Curative or experimental interventions should be discussed early, before complications render the patients ineligible for these treatments.
Introduction
The majority of newborns in well-resourced countries will now survive to adulthood.1,2 In 2017, 23% of all patients reported to the National Haemoglobinopathy Registry in the United Kingdom (nhr.mdsas.com) were ≥40 years of age; in a medium-resourced setting, a follow-up of the Jamaican sickle cell anemia (HbSS) cohort commenced in June 1973 showed that 55.2% have so far lived to 40 years of age.3 In well-resourced settings, estimates of survival varied from the fifth to seventh decade for those with HbSS, and higher for the milder sickle cell disease (SCD) genotypes but these figures were not based on birth cohorts.4,5 For the vast majority of patients who are born in Africa, however, as many as 90% die, usually undiagnosed, during the first 5 years of life.6 Although there is clearly an improvement in survival, the life expectancy of patients with SCD is still shortened by >20 to 30 years compared with the general population. In addition to the acute sickle-related complications, SCD is also associated with multiple chronic organ comorbidities and almost all organ systems can be affected (Table 1; Figure 1).7 Determining causes of death in SCD patients is problematic, not only due to a paucity of autopsy data, but often the final causes of death do not accurately portray the contribution of underlying chronic organ damage. Taken together, studies indicate that the main causes of death in adults (18 years or older) include infection, acute chest syndrome, stroke, renal failure, and pulmonary hypertension.8-11
Commonly recognized sickle-related complications in adults
| Complication | Definition | Major risk factors and prevalence |
|---|---|---|
| Pain | Acute pain, most commonly in the long bones, chest, back | Episodes of acute pain commence from around 6 mo of age and continues throughout life |
| Adults with SCD experience pain on >54% of days but only access health care on 3.5% of days.36 | ||
| Chronic pain is pain lasting for >3 mo | Estimated to occur in >50% adults with SCD; 40% of adults with SCD take daily opioids.99 | |
| Anemia | Acute anemia: a decline in hemoglobin of 2 g/dL or more from steady-state values | Variety of causes including infection (transient red cell aplasia most commonly caused by acute parvo virus B19); acute hemolysis accompanying severe VOC, delayed transfusion reaction |
| Chronic hemolytic anemia: severity increases with age and major contributor to insidious organ dysfunction | Chronic hemolysis predisposes to gallstones and gallbladder disease | |
| ACS | Acute onset of respiratory symptoms with features similar to pneumonia | 24.5/100 PYO in young children; 8.8/100 PYO in older adults101 ; outcome more severe in adults |
| Pulmonary hypertension | Mean pulmonary artery pressure of >25 mm Hg at rest, measured by right heart catheterization | 6.0%-10.4% prevalence during adulthood.41-43,46,47 |
| Cardiac dysfunction | Left ventricular failure is most common abnormality | Universal to some degree in adults over age 30.44 Diffuse myocardial fibrosis is a common pathology, and associated with diastolic dysfunction, anemia, and high NT-proBNP. Fibrosis-mediated diasystolic dysfunction may contribute to elevated TRV in older adults via pulmonary venous hypertension.45 |
| Chronic sickle lung disease | Progressive restrictive lung function deficit with fibrotic changes on high resolution CT scan | Restrictive lung defects seen in >70% adults.38 |
| Asthma | Asthma is seen more commonly in children than in adults | |
| Sleep-disordered breathing | Sleep disordered breathing is seen in 40%-60% of adults.19 | |
| Stroke | Acute cerebrovascular accident | Effective screening and prevention have dramatically reduced infarctive stroke in children. Hemorrhagic stroke affects both children and adults, threefold more in adults.102 Risk factors for ischemic stroke include hypertension, diabetes mellitus, hyperlipidemia, atrial fibrillation, and renal disease in adults.103 |
| Silent cerebral infarcts | Clinically silent lesions of 3 mm or more on magnetic resonance imaging scanning | Important contributing factor to neurocognitive deficits; 53% in adults.20 |
| Acute renal injury | Acute deterioration in renal function | 2% during pain crisis and up to 14% during ACS |
| Renal failure | Deteriorating renal function, reduced concentrating ability, proteinuria, and progressive renal failure | Advanced disease (stage III-IV) in 4%-18% of adults.21 |
| Priapism | Unwanted painful and sustained erection of the penis for >4 h, often recurrent or persistent | 20%-89% lifetime prevalence in boys and men.104 |
| Avascular necrosis of bones | Avascular necrosis of any bone, most commonly the femoral head and shoulder joint | >20% lifetime prevalence of symptomatic disease. Increased prevalence of asymptomatic disease.105 |
| Leg ulceration | Most commonly around the malleolar regions | >14% lifetime prevalence.106,107 |
| Cholelithiasis | Gallstones and gallbladder disease | Important genetic modifier is polymorphic (AT) repeats in promoter of UGT1A1 gene.100 Gallbladder disease in 28% at median age of 28 y.11 |
| Retinopathy | Grade 2-4 retinopathy | >30% of patients.108 Increased in HbSC.109 |
| Complication | Definition | Major risk factors and prevalence |
|---|---|---|
| Pain | Acute pain, most commonly in the long bones, chest, back | Episodes of acute pain commence from around 6 mo of age and continues throughout life |
| Adults with SCD experience pain on >54% of days but only access health care on 3.5% of days.36 | ||
| Chronic pain is pain lasting for >3 mo | Estimated to occur in >50% adults with SCD; 40% of adults with SCD take daily opioids.99 | |
| Anemia | Acute anemia: a decline in hemoglobin of 2 g/dL or more from steady-state values | Variety of causes including infection (transient red cell aplasia most commonly caused by acute parvo virus B19); acute hemolysis accompanying severe VOC, delayed transfusion reaction |
| Chronic hemolytic anemia: severity increases with age and major contributor to insidious organ dysfunction | Chronic hemolysis predisposes to gallstones and gallbladder disease | |
| ACS | Acute onset of respiratory symptoms with features similar to pneumonia | 24.5/100 PYO in young children; 8.8/100 PYO in older adults101 ; outcome more severe in adults |
| Pulmonary hypertension | Mean pulmonary artery pressure of >25 mm Hg at rest, measured by right heart catheterization | 6.0%-10.4% prevalence during adulthood.41-43,46,47 |
| Cardiac dysfunction | Left ventricular failure is most common abnormality | Universal to some degree in adults over age 30.44 Diffuse myocardial fibrosis is a common pathology, and associated with diastolic dysfunction, anemia, and high NT-proBNP. Fibrosis-mediated diasystolic dysfunction may contribute to elevated TRV in older adults via pulmonary venous hypertension.45 |
| Chronic sickle lung disease | Progressive restrictive lung function deficit with fibrotic changes on high resolution CT scan | Restrictive lung defects seen in >70% adults.38 |
| Asthma | Asthma is seen more commonly in children than in adults | |
| Sleep-disordered breathing | Sleep disordered breathing is seen in 40%-60% of adults.19 | |
| Stroke | Acute cerebrovascular accident | Effective screening and prevention have dramatically reduced infarctive stroke in children. Hemorrhagic stroke affects both children and adults, threefold more in adults.102 Risk factors for ischemic stroke include hypertension, diabetes mellitus, hyperlipidemia, atrial fibrillation, and renal disease in adults.103 |
| Silent cerebral infarcts | Clinically silent lesions of 3 mm or more on magnetic resonance imaging scanning | Important contributing factor to neurocognitive deficits; 53% in adults.20 |
| Acute renal injury | Acute deterioration in renal function | 2% during pain crisis and up to 14% during ACS |
| Renal failure | Deteriorating renal function, reduced concentrating ability, proteinuria, and progressive renal failure | Advanced disease (stage III-IV) in 4%-18% of adults.21 |
| Priapism | Unwanted painful and sustained erection of the penis for >4 h, often recurrent or persistent | 20%-89% lifetime prevalence in boys and men.104 |
| Avascular necrosis of bones | Avascular necrosis of any bone, most commonly the femoral head and shoulder joint | >20% lifetime prevalence of symptomatic disease. Increased prevalence of asymptomatic disease.105 |
| Leg ulceration | Most commonly around the malleolar regions | >14% lifetime prevalence.106,107 |
| Cholelithiasis | Gallstones and gallbladder disease | Important genetic modifier is polymorphic (AT) repeats in promoter of UGT1A1 gene.100 Gallbladder disease in 28% at median age of 28 y.11 |
| Retinopathy | Grade 2-4 retinopathy | >30% of patients.108 Increased in HbSC.109 |
ACS, acute chest syndrome; AT, adenine-thymidine; proBNP, N-terminal pro b-type natriuretic peptide; PYO, person-years of observation, VOC, vaso-occlusive crisis.
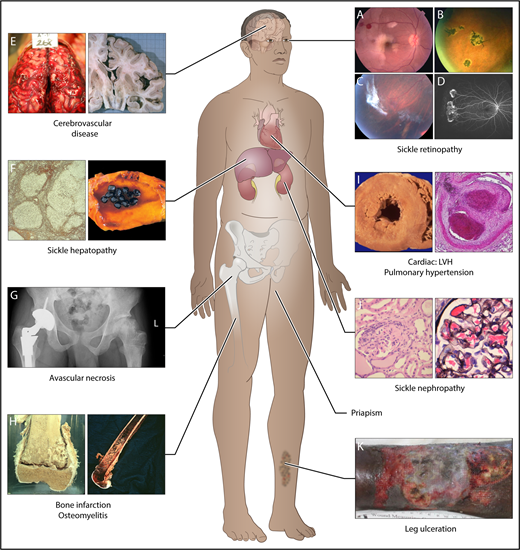
Complications commonly encountered in the older adult with SCD. One study showed that by the fifth decade, almost 50% of surviving SCD patients had documented damage in the organs above.11 In this study, of the 8 organ systems, sickle chronic lung disease was most common, followed by renal failure, retinopathy, osteonecrosis, priapism, gallbladder disease, leg ulcers, and cerebrovascular disease. Frequency of these complications increase with age, but it is also clear that some complications (such as bilirubin levels and gallstones, and sickle nephropathy) are influenced by predisposing or protective genetic variants. The 8 organ system and some of the complications are represented here. (A-D) Sickle retinopathy. Fundus photographs of characteristic retinal lesions observed in sickle cell retinopathy. (A) Arteriolar occlusion with retinal infarcts. (B) Black “sunburst.” (C) Autoinfarcted “sea fan.” (D) Neovascularization in peripheral retina. (E) Cerebrovascular system. Cerebral hemorrhage (left) and atrophy of right cerebral hemisphere (right). (F) Sickle hepatopathy. Light microscopy (silver stain) showing cirrhotic nodules in liver (left) and gallbladder with gallstones (right). (G) Pelvis, avascular necrosis of hip joints. Right hip replacement (left) and early avascular necrosis of left hip (right). (H) Long bones. Infarction of femoral-tibia joint (left) and imaging of osteomyelitis in lower femur (right). (I) Cardiovascular disease. Histopathology showing left ventricular hypertrophy (LVH) of the heart (left) and light microscopy (hematoxylin-and-eosin stain) showing fibrous obliteration of pulmonary vessel with recanalization (right). (J) Sickle nephropathy. Light microscopy (hematoxylin-and-eosin stain) demonstrating large glomerulus and with mesangial hypercellularity (left) and large glomerulus showing sickled red blood cells within the capillaries of the glomerulus (right). (K) Leg ulceration. Chronic intractable ulceration of lower leg above ankle joint.
Complications commonly encountered in the older adult with SCD. One study showed that by the fifth decade, almost 50% of surviving SCD patients had documented damage in the organs above.11 In this study, of the 8 organ systems, sickle chronic lung disease was most common, followed by renal failure, retinopathy, osteonecrosis, priapism, gallbladder disease, leg ulcers, and cerebrovascular disease. Frequency of these complications increase with age, but it is also clear that some complications (such as bilirubin levels and gallstones, and sickle nephropathy) are influenced by predisposing or protective genetic variants. The 8 organ system and some of the complications are represented here. (A-D) Sickle retinopathy. Fundus photographs of characteristic retinal lesions observed in sickle cell retinopathy. (A) Arteriolar occlusion with retinal infarcts. (B) Black “sunburst.” (C) Autoinfarcted “sea fan.” (D) Neovascularization in peripheral retina. (E) Cerebrovascular system. Cerebral hemorrhage (left) and atrophy of right cerebral hemisphere (right). (F) Sickle hepatopathy. Light microscopy (silver stain) showing cirrhotic nodules in liver (left) and gallbladder with gallstones (right). (G) Pelvis, avascular necrosis of hip joints. Right hip replacement (left) and early avascular necrosis of left hip (right). (H) Long bones. Infarction of femoral-tibia joint (left) and imaging of osteomyelitis in lower femur (right). (I) Cardiovascular disease. Histopathology showing left ventricular hypertrophy (LVH) of the heart (left) and light microscopy (hematoxylin-and-eosin stain) showing fibrous obliteration of pulmonary vessel with recanalization (right). (J) Sickle nephropathy. Light microscopy (hematoxylin-and-eosin stain) demonstrating large glomerulus and with mesangial hypercellularity (left) and large glomerulus showing sickled red blood cells within the capillaries of the glomerulus (right). (K) Leg ulceration. Chronic intractable ulceration of lower leg above ankle joint.
There are many unmet needs in SCD,12 particularly in older adults; current management is largely expert consensus-based; evidence-based guidelines are lacking for most complications.13,14 Although pediatric and adult SCD patients share some common phenotypes, the pathobiology and therapy may be age-dependent; we have highlighted certain areas that merit consideration (Figure 2). We have defined the “older adult” as those 40 years of age or older based on current survival estimates; by the age of 40 years, the cumulative organ damage11 will have a significant chronic health burden. We recognize that the rate of development of chronic disease complications is gradual and occurs at variable rates so that much of the evidence and recommendations we give will be applicable to all ages of adults. Observational studies from several centers confirm progressive renal disease and worsening anemia,15-19 as well as incidental and overt neurological complications20 with increasing age. Nonsickle-related comorbidities will also increase with age,18 adding to the disease burden.
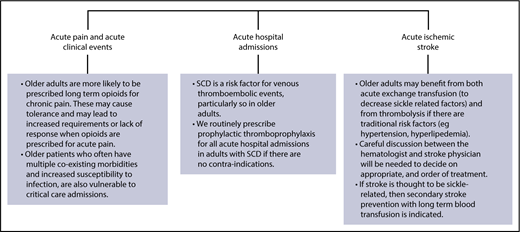
Management of acute complications in older adults with SCD. In general, the management of older adults presenting with acute complications of SCD will not differ significantly from that in younger patients. Note, however, that older adults are more likely to have chronic renal, cardiorespiratory, and/or liver impairment, and may require appropriate dose adjustments of certain medications. Areas where due consideration should be given in older adults include those shown.110-113
Management of acute complications in older adults with SCD. In general, the management of older adults presenting with acute complications of SCD will not differ significantly from that in younger patients. Note, however, that older adults are more likely to have chronic renal, cardiorespiratory, and/or liver impairment, and may require appropriate dose adjustments of certain medications. Areas where due consideration should be given in older adults include those shown.110-113
Here, we present 4 clinical vignettes that highlight some of the particular issues and available therapies when managing the older patient with SCD.
The importance of regular comprehensive review
Case 1
Patient 1 with HbSS presented at age 45 years, feeling tired and unwell. He had significant anemia with hemoglobin (Hb) of 5.4 g/dL. Renal function was severely impaired (chronic kidney disease [CKD] stage 4) with serum creatinine (seCr) of 2.81 mg/dL (normal 0.51-0.95 mg/dL), an estimated glomerular filtration rate (eGFR) of 25 mL per minute per 1.73 m2, and an elevated urinary protein creatinine ratio (uPCR) of 744 mg protein/mmol creatinine. When last seen >5 years ago, his seCr level and eGFR were within normal limits but he did have proteinuria with uPCR of 75 mg protein/mmol creatinine. He declined treatment with an angiotensin-converting enzyme (ACE) inhibitor and was subsequently lost to follow-up.
When he presented again, the patient underwent investigations to exclude other causes of renal dysfunction, including renal tract ultrasound scan and renal biopsy.21 No other cause for renal dysfunction was found; of note, he was normotensive and he was not diabetic. A diagnosis of sickle nephropathy (SCN) was made; he was treated with an ACE inhibitor and hydroxyurea (HU) but his renal function continued to deteriorate, and he was commenced on renal dialysis 1 year later.
Case 2
Patient 2 is a 53-year-old woman with HbSS. At comprehensive annual review, she revealed that although she had not been admitted to hospital during the previous 12 months, she was experiencing pain that was severe enough for self-medication with weak opioid analgesia and to take time off work. Further questioning revealed increasing effort intolerance. She had a long history of snoring.
Cardiorespiratory examination was normal, but her resting pulse oxygen saturations were 92% on air, compared with 95% to 96% on visits in the previous year. Chest radiograph and electrocardiograph were normal; further tests to investigate her shortness of breath included lung function tests, overnight sleep study, computed tomography (CT) chest scanning, and an echocardiograph. The sleep study showed evidence of significant overnight sleep apnea. Assessment by the respiratory physician suggested treatment with continuous positive airways pressure (CPAP) therapy. The patient consented to HU therapy for her repeated acute pain. Six months later, she reported significant decrease in frequency and severity of pain episodes and improved exercise tolerance.
Comments on patients 1 and 2
The overarching principle of care for adults with SCD is regular comprehensive review, sometimes known as the “annual review,” although this may be performed more than once a year (Figure 3). This incorporates a multisystem clinical review, routine blood tests, urine testing and review of other investigations,22,23 assessment of frequency and severity of acute complications, and identification of any sickle-related complications and any other comorbidities, especially those that may interact with SCD.
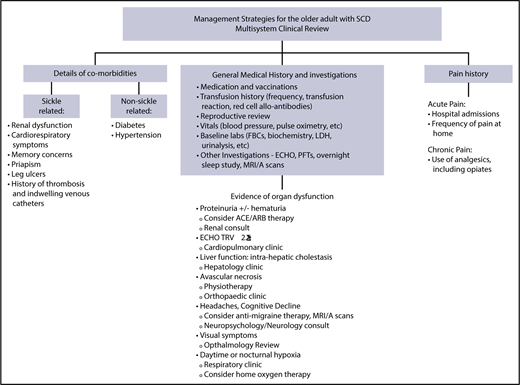
Management strategies for the older adult with SCD. The overarching principle of care for adults with SCD is regular comprehensive review, sometimes known as the “annual review,” using the approach as outlined. During the review, aggregate supportive therapies hydroxycarbamide plus or minus EPO, blood transfusion, and iron chelation therapy, should be discussed and offered if appropriate. The comprehensive review is also an opportunity to provide patient information on new therapies and research studies such as stem cell gene therapy and transplantation.
Management strategies for the older adult with SCD. The overarching principle of care for adults with SCD is regular comprehensive review, sometimes known as the “annual review,” using the approach as outlined. During the review, aggregate supportive therapies hydroxycarbamide plus or minus EPO, blood transfusion, and iron chelation therapy, should be discussed and offered if appropriate. The comprehensive review is also an opportunity to provide patient information on new therapies and research studies such as stem cell gene therapy and transplantation.
Cases 1 and 2 illustrate many of the management strategies for the older adult with SCD (Figure 3).
Comprehensive review initially identified evidence of kidney damage in patient 1. The patient declined specific treatment and was subsequently not seen for several years, by which time the renal impairment had worsened. When he presented again, he was investigated for comorbidities, in particular, hypertension and diabetes, which could accelerate renal impairment. In a cohort of SCD patients over the age of 40 years, 38% had hypertension and 7% had diabetes mellitus.18 Other causes of renal disease, such as lupus nephritis and hepatitis C virus or HIV-associated nephropathy, should also be considered.21 It should be noted that there is no pathognomonic lesion that defines SCN. Treatment of SCN includes a combination of generic measures for ameliorating SCD and its complications, and those interventions that we know to be beneficial in slowing the progression of CKD. For example, aggressive lowering of the systolic blood pressure even to below conventional targets (ie, to <110 mm Hg) has been shown to have additional beneficial effects both on the progression of diabetic nephropathy and on cardiovascular events.24 Patients with SCD tend to have a lower systemic blood pressure for age- and ethnicity-matched controls.25 It has also been shown that patients with SCD and relative hypertension (high-normal blood pressure values of systolic 120-139 mm Hg and diastolic blood pressure 70-89 mm Hg) have both higher seCr levels and an increased risk of pulmonary hypertension when compared with SCD patients with lower blood pressures.26,27 Microalbuminuria (defined as >4.5 mg/mmol) is an early manifestation of SCN, reaching a prevalence of over 50% in older patients.28 In 4% to 12% of the patients, the proteinuria may progress to end-stage renal disease needing renal replacement therapy that is associated with increased mortality.29 Relevant to patient 1, observational data have shown that proteinuria responds to treatment with ACE inhibitors or angiotensin receptor blockers. Although there are no published guidelines on when to introduce ACE inhibitors or angiotensin receptor blockers, all adults should have urinary protein measurements performed at least annually and treatment with these agents should be considered when uPCR is persistently >50 mg/mmol.30,31 Disease-modifying therapy (HU) should be considered alongside this specific treatment.21,32 Both hemodialysis and peritoneal dialysis are effective treatments of end-stage renal failure and choice of dialysis will depend on the patient. The outlook for patients with SCD on dialysis is poor and they have reduced survival compared with other causes of end-stage renal failure33 ; early renal transplantation should be encouraged. Historically (1988-1999), SCD recipients had markedly lower survival compared with other ethnic-matched kidney transplant recipients. In more recent years (2000-2011), patient survival among SCD kidney recipients has greatly improved. At 1-year post renal transplantation, mortality is similar in the SCD and non-SCD population. Six-year patient survival has greatly improved but still trended toward lower survival compared with hypertensive kidney recipients, but there was no difference among matched SCD and diabetic recipients.34,35
A comprehensive review includes taking a thorough pain history and assessment of opiate use. Patient 2 had been experiencing significant pain disrupting her daily life, despite only occasional attendances at hospital, as noted in many SCD patients.36 This may not only impact quality of life but may also lead to significant use of opiate analgesia. Patients who have sickle cell–associated pain that interferes with daily activities and quality of life should be treated with HU.13
Patient 2 also reported increasing effort intolerance; although sickle-related cardiorespiratory complications are common, nonsickle-related causes should also be considered and appropriate investigations performed.37 Up to 90% of adults have abnormalities on pulmonary function testing, the most common being restrictive defects38 and declining forced expiratory volume in 1 second with increasing age.39 Severe chronic sickle lung disease (an abnormality of the lung parenchyma characterized by restrictive lung disease), pulmonary hypertension (PH) (defined as mean pulmonary artery pressure as ≥25 mm Hg at rest diagnosed by right heart catheterization), hypoxia, and chest pain have been described in 5% of SCD patients with an average onset in the third and fourth decades.40 Patients with respiratory symptoms or baseline hypoxia should be investigated with pulmonary function testing and high-resolution CT scanning of the chest. Sleep-disordered breathing is common; both nocturnal hypoxia and overnight sleep apnea have been described in 40% to 60% of adults in small series and a sleep study is recommended in those with suggestive symptoms.19 PH affects 6% to 11% of SCD adults41-43 and is a leading cause of morbidity and early mortality.37 Although left ventricular volume overload and chronic anemia are dominant factors in the abnormal cardiopulmonary hemodynamics,44 it is becoming clear that diffuse myocardial fibrosis is more common than appreciated in SCD, particularly in older adults, and is a key contributor to the diastolic dysfunction.45 Transthoracic echocardiography (ECHO) has been used as a noninvasive tool for screening for PH. Although a raised tricuspid regurgitant velocity (TRV) value of ≥2.5 m/s alone is not a good predictor of PH, it is associated with an increased risk of death and impaired functional status.46,47 TRV values of ≥2.5 m/s identify a group of patients who merit further investigation and perhaps disease-modifying therapy.37,47 We advocate a baseline ECHO in all adults with SCD. Patients with TRV values >2.9 milliseconds or TRV values between 2.5 and 2.9 milliseconds and abnormal 6-minute walk distance (6MWD), or with suggestive symptoms (for example, low pulse oxygen saturation), require further investigations to exclude or confirm PH and/or pulmonary embolism, as these are treatable complications. There is insufficient evidence on the role of sickle-modifying treatment (HU, transfusion) or of targeted PH therapies in SCD. Bosentan was well tolerated in a randomized trial but closed early due to poor recruitment,48 and a trial of sildenafil closed early because of an increase in pain episodes in treated patients.49 Prostacyclin analogs have not been widely used in SCD due to concerns about line thrombosis but retrospective data on 11 patients with SCD and pulmonary arterial hypertension treated for at least 100 days showed that prostacyclin analogs were well tolerated with evidence of functional improvement and no evidence of increased thrombotic events.50 This needs further investigation in prospective trials.
Comprehensive review may also identify other complications which could benefit from intervention, such as joint pain, liver disease, priapism, leg ulcers or visual symptoms. Symptomatic avascular necrosis (AVN) may affect up to 50% of adults with SCD and most commonly affects the hip or shoulder. AVN requires referral to an experienced orthopedic surgeon. Conservative treatment may be appropriate in the early stages; physiotherapy has been shown to be as effective as core decompression.51 Joints that show collapse, and causing significant pain, are likely to need joint replacement. Occasionally, chronic pain may be due to osteomyelitis, which should be treated aggressively with antibiotics. Sickle hepatopathy, an umbrella term encompassing the heterogeneity of liver dysfunction, is more common than appreciated.52 It is important to look for and treat any causes of liver disease outside SCD itself. With the increasing use of blood transfusion, secondary iron overload is an emerging cause of liver disease.
Patients should also be specifically questioned on neurological symptoms. Stroke, seizure, and transient ischemic attacks have been reported in over 30% of adults with SCD, and a history of these events correlates with early mortality.5 The introduction of transcranial Doppler screening and blood transfusion for those at risk has been shown to markedly decrease the incidence of childhood SCD-related stroke but its impact on the incidence of adult stroke is yet to be determined. On reaching older adulthood, patients on long-term transfusion therapy for secondary stroke prevention should continue if alloimmunization and sourcing compatible blood are not an issue. Obviously, such patients require regular monitoring of iron load and chelation therapy as appropriate.
Several studies have confirmed the high rates of intracranial aneurysms in SCD,20,53,54 which is likely to be the cause for the high rate of subarachnoid hemorrhages in adults. An imaging study in 60 asymptomatic adults (median age, 30 years) with SCD found incidental intracranial aneurysms in 9% and silent cerebral infarction in 53% of adults with HbSS and HbSβ0 thalassemia.20 Silent cerebral infarctions have a neurocognitive impact leading to cognitive decline.55 Other sickle-related (anemia, nocturnal hypoxia) and nonsickle-related (hypertension, hyperlipidemia, cardiac disease) factors may also contribute to the accelerated cognitive decline. There is no current consensus guidance on appropriate screening or interventions for diagnosing or treating cognitive decline in SCD but memory concerns should be part of the comprehensive review.56 We do not routinely perform magnetic resonance imaging/angiography (MRI/A) scans in adults but would have a low threshold for this investigation in patients with headaches, poor memory, or other neurological symptoms. Patients with intracranial aneurysms or moya moya should be referred for neurosurgical review.
Needless to say that when patients have a long period of stability and present with worsening of their sickle phenotype, the clinician should always be aware of new comorbidities as an explanation.18
The comprehensive review is also an opportunity to provide patient information on new therapies, and to engage the patient in his/her own care. This may include discussion of the need for vaccination and infection prophylaxis and discussion of reproductive plans (if appropriate) including a reminder of the need for partner screening and discussion of contraception if required.
The role of disease-modifying therapy in older adults
Case 3
Patient 3 is a 54-year-old woman diagnosed with HbSS at 8 years of age. Between age 10 and 20 years, she had minimal symptoms, few ER visits and no hospital admissions. From age 20 years, she started experiencing increasing acute pain needing hospital admissions, occasionally requiring red blood cell (RBC) transfusions during this period. From age 40 years, she averaged 3 to 4 admissions per year, with 1 admission at age 46 years that lasted a whole month. In between the hospital admissions, she had an average of 30 mild/moderate pain crises per year. In addition to the episodic acute pain, she had chronic leg pain that was moderately severe, related to a prior left femur surgery with metal rod placement at age 20 years. She has received >50 units of RBC in her lifetime, and she has known RBC alloantibodies.
She was commenced on HU therapy at age 35 years, which was stopped within a few months when she became pregnant. HU was recommenced 8 years later, but was eventually stopped after 2 years due to lack of clinical response.
The patient had a fully matched HbAA sibling and was considered for hemopoietic stem cell transplant (HSCT). Pre-transplant evaluation noted: left parietal occipital infarct on brain MRI; borderline TRV of 2.5 to 2.6 m/s with right ventricular systolic pressure of 30 to 33 mmHg and normal ejection fraction of 65%; iron overload (liver iron concentration, 16mg/g DW); proteinuria with albumin-creatinine ratio of 130 to 530 mcg/L; bilateral AVN of shoulders and knees.
She underwent nonmyeloablative sibling donor HSCT at age 48 years. Her hematologic parameters improved with latest chimerism of 44% CD3 and 100% CD14/15, Hb 13 g/dL (from pretransplant baseline Hb 7-8 g/dL). There were also improvements in her pulmonary function testing and 6MWD. Her ECHO, brain MRI, and renal function remained stable, whereas liver function has completely normalized with normal bilirubin and LDH levels. With older age and weight gain, her BP became high to the point where she needed 2 medications for BP control. She is free of acute sickle pain but takes Percocet (a combination of oxycodone and acetaminophen) for arthritic pain in her knees. She is also on regular phlebotomy as treatment of her iron overload.
Case 4
Patient 4 is a 40-year-old woman with HbSS with numerous complications: recurrent lower extremity ulcers, osteomyelitis, multiple episodes of acute chest syndrome, multiorgan system failure (secondary to severe acute pain crisis on 1 occasion, and severe delayed transfusion reactions on 2 occasions), sickle-related chronic kidney disease, cholelithiasis and cholecystectomy, secondary iron overload treated with iron chelation, cardiomyopathy with systolic and diastolic dysfunction, severe life-threatening anemia, and sickle hepatopathy. She has multiple RBC alloantibodies.
Her previous sickle related therapies included HU, repeatedly interrupted due to poor healing of her cutaneous leg ulcers; subsequently, HU was stopped due to poor clinical response. Attempts to improve her hemoglobin levels included decitabine as an investigative drug which was discontinued due to thrombocytosis and lack of improvement in hemoglobin levels. A severe painful crisis at age 36 years triggered an acute chest syndrome and her first of several episodes of multiorgan failure that required hemodialysis for acute renal failure on one occasion. Since then, her baseline hemoglobin hovers around 5 g/dL. Her severe anemia has also been treated intermittently with erythropoietin (EPO) over the last 15 years, but EPO therapy appeared to trigger acute painful episodes.
Blood transfusion was always complicated by delayed reactions triggering further acute pain. Subsequent serial testing revealed warm autoantibody with preference for RH and anti-Cw alloantibody despite previously receiving C-negative blood. Molecular testing and DNA sequencing revealed normal RHD alleles and heterozygosity for RHCE*ce254C>G allele. RBC transfusion remained a major problem, only “least incompatible bloods” were available despite extended red cell antigen matching of the transfused units. IVIG and steroid cover did not help, she could not maintain her hemoglobin that often dropped to pretransfusion levels, and on 2 occasions, Hb levels fell to <2 g/dL. Withdrawal of steroid cover further triggered VOCs.
A joint decision was made with the patient to withhold RBC transfusion unless absolutely life-saving. She remains transfusion free and is not receiving traditional disease modifying therapy.
Comments on patients 3 and 4
Long-term HU has been the only disease-modifying drug available until recently, but it is not effective in all cases. Regular RBC transfusion provides an alternative, but alloimmunization to RBC antigens can lead to difficulties in sourcing appropriately matched blood products, as in patient 4. In those patients in whom neither HU nor regular blood transfusion are possible or effective, other interventions should be considered. This may include HSCT (as offered to patient 3), gene therapy or experimental drug therapy.
Disease-modifying therapy in adults with SCD
Hydroxyurea
Until July 2017, when Endari (l-glutamine oral powder) was licensed by the FDA, HU was the only licensed drug for treating SCD. Evidence for its efficacy in reducing acute pain, acute chest syndrome and transfusion needs,57 has led to its expanded use over the last decade. Longitudinal studies of >5 years in different international cohorts has shown improved survival in those patients taking long-term HU although the studies were not randomized.58-62 Early instigation of HU prevents acute complications and improve childhood survival61,63 but it is still not clear if long-term hydroxyurea therapy will prevent organ damage, and ultimately, improve long-term survival in adulthood. Initiation of HU in very young children did not show a difference in splenic function or in glomerular function rate, although treated patients did show better urine concentrating ability and less renal enlargement.63,64 In adults, observational data suggested that HU may improve renal dysfunction and albuminuria, but this needs replication in a randomized trial.32 A randomized controlled trial in pediatric population concluded that HU was comparable to blood transfusion for primary stroke prevention,65 but not in secondary stroke prevention.66 Again, these results may not necessarily translate directly to adults with SCD.
Recently, the relative safety of HU in settings with high infectious disease burden have increased our confidence in broadening the clinical use of this drug.67 Although the role of HU in the prevention of chronic sickle complications needs to be verified in randomized controlled trials, there is sufficient evidence of its impact on survival and acute complications to conclude that, at the very least, HU therapy should be discussed with adults with all SCD genotypes at the comprehensive annual review. HU does have a long-term safety profile and minimal side-effects, and many care providers argue that SCD patients should be offered HU treatment early rather than when they present with severe complications. Despite the clear benefits of HU, it remains underutilized, probably due to a reluctance in both patients and clinicians.68 Long-term HU treatment starting in childhood may produce a generation of adults with less acute complications, fewer hospital admissions, decreased organ damage and improved survival. Conversely the effects of hydroxyurea on fertility (especially in prepubescent boys) are poorly understood and we may be facing complex fertility issues in our adults in the future.
We have observed that the older SCD patient is very sensitive to HU dosage, with increased rates of neutropenia, thrombocytopenia and reticulocytopenia. HU therapy therefore needs careful and more frequent monitoring in older adults compared with younger patients. It has been suggested that repeated bone marrow infarcts over the years and reduced hemopoietic potential contributes to the relatively increased HU sensitivity in the older patient.
Long-term blood transfusion therapy
Although chronic RBC transfusion has an evidence base for stroke prophylaxis or treatment in children, its use in many other medical situations, such as prevention of recurrent acute pain, priapism, leg ulcers, renal dysfunction and pulmonary hypertension, remains pragmatic based on observations and clinical experience. Evidence for the efficacy of transfusion in preventing pain, acute chest syndrome and other complications is from secondary outcome data from randomized trials in pediatric stroke prevention.69 The aim of long term transfusion is to maintain a relatively low concentration HbS%. The HbS% target will vary according to indication, we aim for a HbS% of <30% for primary or secondary stroke prevention but when prescribed for limitation of recurrent acute pain, or to hasten healing of recalcitrant leg ulceration, a relatively higher concentration of HbS% <40% to 50% may suffice. For other indications, such as managing sickle cell intrahepatic cholestasis, suggested thresholds have been reported as <20 and <30%.52,70,71 Long term blood transfusion therapy can be given as repeated simple transfusion or as exchange transfusion using manual or automated methodology. The former can be used for patients with marked anemia but results in iron loading and may not adequately suppress the HbS%. Both automated and manual RBC transfusion will maintain an appropriate HbS% with minimal iron loading, but economic analysis favors the former.72
Given the global clinical benefits, it is not surprising that the use of long term blood transfusion therapy in the management of adults with SCD is increasing. A single center study from the UK showed that the number of adults receiving long term blood transfusion increased from 15% to 19%, with an increase from 1.7 to 3.86 number of units administered per patient year over a ten year period.73
A serious complication of blood transfusion is alloimmunization to RBC antigens, which is relatively more common in patients with SCD,74-77 and can lead to difficulties in sourcing appropriately matched blood products, delayed transfusion reactions triggering more VOCs, significant morbidities, and fatalities in some cases.75,78-81 Despite ethnic matching, a significant proportion of SCD patients still become alloimmunized.77 Molecular analyses of the RH genes in SCD patients and African American donors revealed remarkable RH allelic diversity in this population and mismatch between serologic Rh phenotype and RHD or RHCE genotype owing to variant RH alleles in 87% of individuals,77 as encountered in patient 4. As a prevention, many blood transfusion centers have adopted extended red cell phenotyping, including ABO, Rh, Kell, Kidd, Duffy, and S and s antigens, and some centers have also adopted molecular genotyping for red blood cell phenotype prediction using microarray chips (eg, the PreciseType HEA BeadChip assay).
Long term blood transfusion also leads to iron overload and difficulties of obtaining adequate venous access. In SCD patients, secondary iron overload most commonly affects the liver, cardiac and endocrine impairment are unusual. Patients treated with sporadic blood transfusions can also develop a substantial iron overload over time, which may go unnoticed.82,83 Magnetic resonance imaging (R2MRI) is a noninvasive method of measuring body iron load and should be used in those with a high annual blood intake and raised ferritin.84 Iron chelation therapy should be considered in those with serum ferritin >1000ug/l and liver iron concentration of >7mg/g dry weight and efficacy measured with serial serum ferritin and R2MRI measurements. For those patients on long term transfusion therapy, automated exchange transfusion is associated with reduced iron loading compared with manual exchange transfusion or simple transfusion therapy.85
Hemopoeitic stem cell transplant
HSCT is a curative option for patients with SCD. One thousand HLA-matched sibling HSCTs performed between 1986 and 2013 reveal excellent outcomes for both children and adults demonstrating 93% overall survival (95% confidence interval, 91.1% to 94.6%).86 However, outcomes were poorer in those over 16 years (n = 154). Reduction of the intensity of conditioning (nonmyeloablative) has expanded allogeneic transplantation as a treatment option for adult patients with preexisting organ dysfunction, who would have been otherwise ineligible for transplantation using standard myeloablative conditioning.87 The HLA-matched sibling donors can be HbAS or HbAA, as long as donor myeloid chimerism is >20% which is adequate to reverse the sickle hematological phenotype.88,89 However, in the US, less than 15% of patients with SCD have HLA-matched siblings as donors.90 Hence, several approaches are needed to make HSCT available to more patients, and these include expanding donor sources from which stem cells can be obtained, such as an umbilical cord blood and haploidentical family members or matched unrelated donors, combined with less intensive conditioning strategies and better supportive care.91 Of these alternative donor sources, the most promising is haploidentical family members.92,93
New therapies
Successful curative treatment with additive (anti-sickling β-globin vector containing the HbAT87Q mutation (Bluebird Bio)), gene therapy has recently been described,94 albeit in a single patient, and is currently being investigated in several clinical trials internationally. The last 2 decades, has also witnessed a surge in the development of drugs that target HbS polymerization,95 and those that prevent or reverse vaso-occlusion.96 Approaches to preventing HbS polymerization include increasing fetal hemoglobin via both gene therapy and pharmacologically. A large number of drugs with various modes of action are currently in phase 2 and 3 trials. From the perspective of preventing acute pain, crizanlizumab, a humanized antibody to P selectin holds much promise. In a double-blind, randomized, placebo-controlled phase 2 trial, adult participants (on or off HU), Crizanlizumab showed a significant reduction in sickle related pain crises and low incidence of adverse events.97 A phase 2 trial has shown efficacy of a pan-selectin inhibitor, rivipansel, in the treatment of acute crises and is currently undergoing phase 3 investigation (http://www.clinicaltrials.gov/, #NCT01119833).98 A hemoglobin modifier (Voxelotor) is also currently being investigated in a phase 3 trial (https://www.clinicaltrials.gov/, NCT03036813; NCT02850406). Voxelotor modulates hemoglobin affinity for oxygen and by maintaining a proportion of hemoglobin S in the oxygenated state, it inhibits HbS polymerization and the resultant sickling of red cells.
Conclusion
Limited resources and data on health care issues pose multiple challenges for managing the older patient. Disease-modifying therapies (HU and blood transfusion) should be considered and offered to the older patient if they are eligible as should curative therapy with HSCT and (potentially) gene therapy. Patients do best when managed in a specialist clinic by a core multidisciplinary team with co-ordinated review by multiple specialists and supportive input from allied health professionals, including psychology and physiotherapy. This guideline is written from a perspective of a well-resourced setting. The reality of most resource-strained countries is very different, and the health provider has to appropriate the best approaches for managing not just adults, but all patients with SCD in their own settings. Although we define “older” as 40 years or more, there is no timeline for these complications: the onset and rate vary greatly between patients.
Acknowledgments
The authors thank Matt Hsieh (Sickle Cell Branch, National Heart, Lung, and Blood Institute, National Institutes of Health) for sharing clinical vignette 3. The authors also thank Rusinel Amarante for help in preparation of the manuscript.
This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health (S.L.T.).
Authorship
Contribution: S.L.T. and J.H. cowrote the paper.
Conflict-of-interest disclosure: J.H. has received honoraria from Novartis, Terumo BCT, AddMedica, and GBT440. S.L.T. declares no competing financial interests.
Correspondence: Swee Lay Thein, Sickle Cell Branch, National Heart, Lung and Blood Institute, National Institutes of Health, Building 10-CRC, Room 6S241, 10 Center Dr, Bethesda, MD 20892; e-mail: sl.thein@nih.gov.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal