

Abstract
The incidence of venous thromboembolism (VTE) in adult patients with sickle cell disease (SCD) is high. However, overlapping features between the clinical presentation of VTE and SCD complications and a low index of suspicion for thrombosis can influence patient management decisions. VTE in SCD can therefore present management challenges to the clinical hematologist. Herein, we present 3 distinct clinical vignettes that are representative of our clinical practice with SCD patients. These vignettes are discussed with specific reference to the hypercoagulable state in SCD patients, recent VTE diagnosis and anticoagulant therapy guidelines from the general population, and evaluation of the risk of bleeding as a result of long-term exposure to anticoagulant therapy. We examine current diagnostic and treatment options, highlight limitations of the existing clinical prognostic models that offer personalized guidance regarding the duration of anticoagulation, and propose a clinical approach to guide the decision to extend anticoagulation beyond 3 months.
Introduction
Sickle cell disease (SCD; herein defined as homozygous hemoglobin [Hb] SS, or compound heterozygous HbS/β-thalassemia or HbS/HbC) is the most common inherited blood disorder in the United States, and a major health problem throughout the world.1 Over the last 5 decades, greater disease awareness by patients and improved access to care has reduced overall morbidity and mortality of SCD, especially in high-income countries.2,3 New medical therapies are being developed at a faster pace, and hematopoietic cell transplantation and somatic gene therapy offer curative potential.4-6 Increased life expectancy in adults with SCD is leading to a greater appreciation of organ complications.
SCD has long been considered a disorder primarily of erythrocytes wherein abnormal polymerization of Hb tetramers upon deoxygenation results in intermittent painful episodes, hemolytic anemia, vascular inflammation, and vaso-occlusion, eventually compromising organ function. Hypercoagulability, defined by many biomarkers that denote activation of prothrombotic factors or decreased antithrombotic proteins, is well described in patients with SCD.7,8 The contribution of hypercoagulability to the pathophysiology of common complications (vaso-occlusive crisis [VOC], stroke, acute chest syndrome [ACS]) of SCD is uncertain and therapeutic trials of anticoagulant drugs or platelet inhibitors have shown conflicting results.9
Venous thromboembolism (VTE), defined as deep vein thrombosis (DVT) or pulmonary embolism (PE), is increasingly recognized as a frequent and important clinical complication in adults with SCD, and is likely, at least in part, the result of this hypercoagulable state.10-12 In these reports, up to 12% of patients with SCD have a VTE by 40 years of age.12 Moreover, the VTE recurrence rate in SCD patients is similar to those individuals in the general population with unprovoked VTE, and is associated with increased mortality.10-12 There is no evidence from randomized trials that the management of SCD patients with VTE should be different from that recommended for other adults. However, within the prevailing paradigm, there are unanswered questions. Should SCD, in and of itself, be considered a strong persistent underlying risk factor for recurrent VTE warranting indefinite anticoagulation after a single incident VTE? Alternatively, should SCD be considered a mild thrombophilia, with a shorter duration of secondary pharmacological prophylaxis and further therapy only during exposure to periods of higher risk? How is the clinical paradigm of provoked and unprovoked VTE applicable to this population? Finally, are patients with SCD and VTE at increased risk of bleeding?
We believe that carefully designed randomized clinical trials to identify appropriate primary and secondary prevention strategies for VTE in SCD patients are warranted, given the frequency of this complication in adults and its contribution to mortality. Compared with VTE in the limbs or pulmonary vasculature, the frequency and importance of risk factors associated with VTE in unusual locations (eg, cerebral sinus thrombosis) are possibly different and beyond the scope of this article. In addition, we will not discuss primary VTE prophylaxis for the hospitalized SCD patient, other than propose that such patients be given pharmacological prophylaxis considering their high risk for VTE.
Absent direct evidence, clinicians are left with making management decisions based on extrapolations of general VTE treatment paradigms to SCD patients.13 In the current article, we discuss 3 commonly encountered case scenarios of VTE in SCD in our practices to illustrate how we diagnose and manage this problem. Our goal is to enable hematologists caring for SCD patients to be able to: (1) understand the hypercoagulable state in SCD and quantify VTE risk, (2) discuss the type and duration of anticoagulation for an incident VTE event in SCD patients, and (3) identify situations that warrant extending anticoagulation beyond that required for active treatment of VTE in SCD, weighing the risk of recurrence against that of major bleeding.
Case 1: acute DVT
A 42-year-old African American man with HbSS presents to the hospital with acute-onset left leg swelling. He has no previous history of venous thromboembolic disease, had not been hospitalized for 2 years, and had no recent operations. His mother, who did not have SCD, had an idiopathic lower-extremity DVT at 51 years of age. He has no cardiopulmonary symptoms. D-dimer was elevated, and bilateral Doppler ultrasound revealed an acute occlusive venous thrombosis of the left femoral vein extending from the popliteal trifurcation to the iliac vein. The patient was administered rivaroxaban at a dose of 15 mg orally, twice daily, for 21 days, and then reduced to 20 mg once daily.
Case 2: pregnant SCD patient with a history of DVT
A 32-year-old G1P0 Ghanaian woman with HbSS is 10 weeks pregnant. She developed a left femoral vein deep venous thrombosis at age 26 years during a hospitalization for VOC, and was treated with low-molecular-weight heparin (LMWH) followed by warfarin for 6 months. She has not had recurrent VTE. You are asked to make recommendations for VTE prophylaxis during pregnancy.
Case 3: catheter-related upper-extremity thrombus and its management
A 21-year-old African American woman with SCD on chronic, monthly exchange transfusion therapy for a history of ischemic stroke at 12 years of age presents with sudden-onset pain and swelling of her right upper extremity. She has a double-lumen port-a-cath in the left chest with entry through the left subclavian vein that was placed 2 years ago. She has a progesterone-eluting intrauterine device in place for a year. A Doppler ultrasound reveals an occluding thrombosis of the right axillary, subclavian, and internal jugular vein. Urine pregnancy test is negative, and she is placed on rivaroxaban.
The biochemical and clinical evidence for a hypercoagulable state in SCD
Hypercoagulability in the pathophysiology of SCD
Although HbS polymerization, hemolytic anemia, and impaired microcirculatory blood flow from acute vaso-occlusion are central to disease pathophysiology, combined together they precipitate a cascade of downstream pathologic events that seemingly lead to organ complications and thrombotic vasculopathy (Figure 1). The contribution of coagulation activation, inflammation, and ischemia/reperfusion to the vascular pathobiology of SCD has been recently summarized.14-16
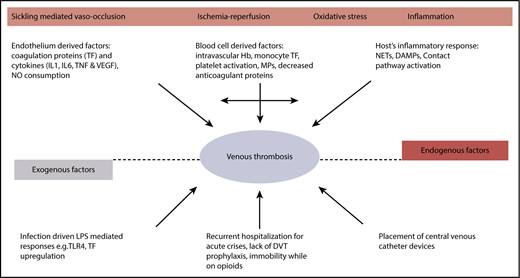
Hypercoagulability in SCD. DAMP, damage-associated molecular pattern; IL, interleukin; LPS, lipopolysaccharide; MP, microparticle; NET, neutrophil extracellular trap; TF, tissue factor; TLR, Toll-like receptor; TNF, tumor necrosis factor α; VEGF, vascular endothelial growth factor.
Hypercoagulability in SCD. DAMP, damage-associated molecular pattern; IL, interleukin; LPS, lipopolysaccharide; MP, microparticle; NET, neutrophil extracellular trap; TF, tissue factor; TLR, Toll-like receptor; TNF, tumor necrosis factor α; VEGF, vascular endothelial growth factor.
There is abundant biomarker evidence for ongoing activation of the coagulation cascade both during steady state (clinically well) and during VOC (Table 1). This may be triggered by the increased expression of tissue factor on monocytes and endothelial cells in the circulation of patients with SCD.17-21 Procoagulant protein activation is further accelerated by phosphatidylserine exposure on platelet and erythrocytes,22,23 and cell-derived microparticles,21,24-27 which serve as a surface for cell-based thrombin generation.28 Cell-free heme, increased in SCD from hemolysis, can induce endothelial tissue factor expression.29 In addition, arginase I released from the red blood cells during hemolysis depletes arginine, which is the substrate for nitric oxide synthesis.30 The resultant decrease in nitric oxide further tilts the hemostatic balance toward thrombosis.
Alterations in the coagulation system in humans with SCD
| Biochemical evidence of coagulation activation | Increased levels | Decreased levels | Steady state | Acute crisis | Reference |
|---|---|---|---|---|---|
| FXa generation | |||||
| Tissue factor pathway | + | + | + | 18, 68 | |
| Intrinsic pathway (FXII, HMWK, prekallikrein) | − | − | 31, 32 | ||
| Thrombin/fibrin generation | |||||
| D-dimer | + | + | + | 69 | |
| Prothrombin fragment F1.2 | + | + | + | 70 | |
| Fibrinopeptide A | + | 71 | |||
| Thrombin antithrombin complexes | + | + | + | 68 | |
| Changes in anticoagulant protein levels | |||||
| Protein C and S | − | − | − | 72, 73 | |
| ATIII and heparin cofactor II | − | − | 74, 75 | ||
| Changes in fibrinolytic protein levels | |||||
| Plasminogen activator inhibitor 1 | + | + | + | 76, 77 | |
| Other plasmatic factors | |||||
| FVIII | + | + | 78 | ||
| VWF | + | + | 79 | ||
| Cellular factors contributing to coagulation | |||||
| Platelet number and size | + | + | * | 22 | |
| Platelet activation and function | + | + | + | 80 | |
| Red blood cell activation (PS exposure and adhesion) | + | + | 81 | ||
| Leukocyte activation (TF exposure) | + | + | + | 20 | |
| Endothelial activation (TF exposure and adhesion) | + | + | + | 17 | |
| Cell-derived microparticles | + | + | + | 21 |
| Biochemical evidence of coagulation activation | Increased levels | Decreased levels | Steady state | Acute crisis | Reference |
|---|---|---|---|---|---|
| FXa generation | |||||
| Tissue factor pathway | + | + | + | 18, 68 | |
| Intrinsic pathway (FXII, HMWK, prekallikrein) | − | − | 31, 32 | ||
| Thrombin/fibrin generation | |||||
| D-dimer | + | + | + | 69 | |
| Prothrombin fragment F1.2 | + | + | + | 70 | |
| Fibrinopeptide A | + | 71 | |||
| Thrombin antithrombin complexes | + | + | + | 68 | |
| Changes in anticoagulant protein levels | |||||
| Protein C and S | − | − | − | 72, 73 | |
| ATIII and heparin cofactor II | − | − | 74, 75 | ||
| Changes in fibrinolytic protein levels | |||||
| Plasminogen activator inhibitor 1 | + | + | + | 76, 77 | |
| Other plasmatic factors | |||||
| FVIII | + | + | 78 | ||
| VWF | + | + | 79 | ||
| Cellular factors contributing to coagulation | |||||
| Platelet number and size | + | + | * | 22 | |
| Platelet activation and function | + | + | + | 80 | |
| Red blood cell activation (PS exposure and adhesion) | + | + | 81 | ||
| Leukocyte activation (TF exposure) | + | + | + | 20 | |
| Endothelial activation (TF exposure and adhesion) | + | + | + | 17 | |
| Cell-derived microparticles | + | + | + | 21 |
+, increased compared with controls; −, decreased compared with controls; ATIII, antithrombin III; FVIII, factor VIII; FXa, factor Xa; FXII, factor XII; HMWK, high-molecular-weight kininogen; PS, phosphatidylserine; TF, tissue factor; VWF, von Willebrand factor.
Variable findings.
Proximal intrinsic pathway coagulation protein alterations have also been reported in patients with SCD.31-33 Because components of the contact system are mediators of inflammation, activation of this system might play a role in inflammatory pathway perturbations that contribute to a prothrombotic state. Decreases in anticoagulant proteins, such as proteins C and S, have been reported in SCD and likely contribute to the hypercoagulable state.8,14 Finally, the possible role for iron in hypercoagulability has been suggested by decreased ex vivo measures of clotting, as measured by thromboelastography, in the plasma of SCD patients after iron chelation.34
Platelet activation may further promote clot formation. Increased expression of P-selectin on circulating platelets, and plasma soluble factors 3 and 4, β-thromboglobulin, and platelet-derived soluble CD40 ligand are all evidence of ongoing platelet activation in SCD patients.8,14 The clinical relevance of hemostatic activation is suggested by evidence that hydroxyurea treatment lowers many of these markers of hemostatic activation. Therefore, some of the benefit from hydroxyurea in SCD may be through attenuating the hypercoagulable state.35
Recent evidence for increased venous thrombotic events in SCD
For decades, clinicians have suspected that patients with SCD were at an increased risk for VTE but the data were primarily based on limited case series, single-institution studies, and/or case-control studies often confounded by risk factors in the general population.10,36-38 Recently, 2 large retrospective studies, 1 using a natural history cohort and the other using an administrative database from the state of California, have carefully described the incidence of VTE and its sequelae in SCD patients. Retrospectively analyzing data from the Cooperative Study of Sickle Cell Disease (CSSCD) to calculate incidence rates for first-time VTE, Naik and colleagues found an incidence rate of 5.2 events per 1000 person-years (95% confidence interval [CI], 3.8-6.9) in 1523 SCD patients aged ≥15 years with 8862 years of follow-up, with a cumulative incidence of 11.3% (95% CI, 8.3-15.3) by age 40 years.11 Individuals with SS or Sβ0-thalassemia had the highest rate of VTE (7.6 events per 1000 person-years [95% CI, 5.3-10.6]). These incidence rates are comparable with VTE incidence rates observed in prospective cohort studies of patients with inherited thrombophilia. Furthermore, the risk for death of SCD patients with VTE was higher than in those without VTE (adjusted hazard ratio [HR], 2.32; 95% CI, 1.20-4.46). The incidence of PE exceeded that of isolated DVT (3.6 events per 1000 person-years [95% CI, 2.5-5.1] vs 1.6 events per 1000 person-years [95% CI, 0.9-2.7]), although this difference was not statistically significant.
Similarly, Brunson and colleagues, using a population-based administrative database from the state of California, found that by age 40 years, the cumulative incidence of VTE among all SCD patients was 12.5% (95% CI, 11.5-13.6).12 Fifty-two percent presented as PE (±DVT), 25% isolated lower-extremity DVT, and 23% as isolated upper-extremity DVT. Overall, 60% of the VTE events occurred ≤90 days of a prior inpatient hospital discharge, with 94% of these with antecedent inpatient admission lasting >3 days. Among SCD patients, nonpregnant women (HR, 1.18; 95% CI, 1.01-1.38) and those with severe disease (defined as an average of 3 or more hospitalization per year) had an increased risk of VTE (HR, 2.86; 95% CI, 2.42-3.37). Among patients with severe SCD, the 5-year recurrence rate was 36.8%. Overall, VTE was associated with an increased risk of death (HR, 2.88; 95% CI, 2.35-3.52). Taken together, these 2 large studies demonstrated an increased rate of thrombotic events in SCD patients and the similarity of the estimates for incidence and VTE-related mortality are reassuring regarding robustness of the results.
Pregnancy is a well-established risk factor for VTE for women but this risk is magnified in pregnant women with SCD. In 1 study, SCD was an independent risk factor for pregnancy-related VTE with an odds ratio of 6.7 (95% CI, 4.4-10.1).39 Seaman and colleagues examined inpatient hospital discharge data from 212 hospitalized deliveries in African American women with SCD: 6 (2.8%; 95% CI, 1.0%-5.9%) had VTE compared with 0.05% to 2.0% in the general population.40 Overall, the prevalence of VTE among hospitalized deliveries in SCD women with pneumonia, VOC, and/or ACS was significantly greater than among those without these conditions (6.6% vs 2.2%; P < .001). They concluded that pregnancy-related VTE in women with SCD appears to be 1.5 to 5 times greater than pregnancy-related VTE in the general population. Porter and colleagues examined the relationship between sickle hemoglobinopathies and VTE risk during pregnancy or the puerperium.41 Of 103 women with HbSS, HbSC, or HbSβ-thalassemia, 3 women (2.9%) experienced VTE. Compared with women with normal Hb status, the relative risk was 32.2 (95% CI, 9.7-107). The relationship between sickle cell trait, SCD, and VTE in pregnancy has recently been reviewed.42
In the management of pregnancy and SCD, some groups suggest low-dose aspirin as prophylaxis against preeclampsia, and consideration of LMWH when additional risk factors are present. These risk factors include but are not limited to previous VTE, family history of VTE, known thrombophilia, older age, obesity, severe varicose veins, preeclampsia, immobility, and frequent hospitalization.43 Related to pregnancy is the use of oral contraceptives in women with SCD, which is reviewed elsewhere.44-46 Suffice it to say that progesterone-only methods of contraception are the least thrombogenic and are routinely considered first-line.44
Children with SCD are also affected by VTE. Using the Pediatric Health Information System database to investigate all pediatric patients with SCD admitted to 48 participating institutions between January 2009 and September 2015, Kumar and colleagues identified index VTE events and chronic medical conditions known to be associated with VTE using billing codes.47 Of 10 454 eligible subjects with SCD identified, 181 (1.7%) developed an index VTE event at a median age of 15.9 (±7.4) years. On multivariable logistic regression analysis, central venous catheter placement, chronic renal disease, history of stroke, female sex, length of hospitalization, admission to the intensive care unit, and older age were associated with VTE. After adjusting for other variables, VTE was independently associated with death.
Other clinical manifestations potentially involving hypercoagulability in SCD
Recent studies demonstrate the association of thrombosis in situ in the large pulmonary vessels in ∼20% of patients diagnosed with ACS, and post mortem studies of SCD patients diagnosed with ACS showing thrombi in the small pulmonary vessels support a role for thrombosis in disease pathophysiology.48-50 However, the nature of these studies makes it hard to ascertain a cause-effect relationship; that is, whether the presence of thrombosis was a primary ACS-inciting event or whether it occurred as a result of ACS. Results from an ongoing trial of anticoagulation in ACS patients (NCT02580773) could provide evidence or lack thereof regarding the impact of thrombosis on this complication.
Diagnosis of VTE in SCD
Risk factors associated with development of VTE
As with any patient in whom a diagnosis of VTE is being considered, the pretest probability of disease should be assessed. As previously noted, the diagnosis of SCD is an ongoing risk factor for VTE. VTE risk varies with genotype, with HbSS and Sβ0-thalassemia patients having the highest risk compared with HbSC or Sβ+-thalassemia,11 although the same investigators reported a higher incidence with other genotypes when studying a smaller, single-institution cohort of patients.10 Patients who have undergone splenectomy for other diseases,51 especially hemolytic anemias, have increased risk of VTE,52 and this is also likely for SCD patients. SCD patients averaging more than 3 admission per year, and women (even when accounting for pregnancy) were at higher risk for VTE.12,47 As noted previously, 60% of the California cohort were hospitalized within 90 days of incident VTE.12 Those with an elevated tricuspid regurgitant jet velocity on cardiac echocardiography were also at increased risk for VTE in the cohort series from Johns Hopkins.10 Upper-extremity VTE also occurs frequently in patients and is often associated with the presence of an indwelling catheter,10,12,47 suggesting that the risks-to-benefit ratio of indwelling catheter placement in SCD patients must be weighed carefully.
Symptoms and signs of VTE may overlap with other clinical complications of SCD. Lower-extremity edema might be attributed to right heart failure, kidney, or liver disease but its unequal distribution may provide a clue indicating VTE. A unilateral painful, swollen leg might be attributed to cellulitis, bony infarct, or complication of leg ulcers. Shortness of breath, pleuritic chest pain, fever, and hypoxemia are often attributed to ACS and/or pneumonia. Pulmonary emboli may be also associated with wheezing and pulmonary infiltrates. Therefore, a high clinical suspicion for VTE in patients with SCD with a low threshold for diagnostic evaluation is recommended.
Diagnostic testing
There are no studies that directly address the diagnostic algorithm for VTE in patients with SCD but we follow the algorithm shown in Figure 2. The use of D-dimer levels in diagnosis is uncertain, given baseline elevations even when patients are clinically well.8,14 In the general population, D-dimer testing, particularly when adjusted for age, can guide clinical decisions because of its high negative predictive value to rule out VTE and when tested serially, could help individualize the decision to extend anticoagulation duration.53 Normal D-dimer levels may be useful to rule out VTE, but it is rather unusual in our experience, and if the patient has a high pretest probability, one usually proceeds to imaging studies. D-dimer testing does not have a clear role in diagnosis or treatment of VTE in SCD and further research could clarify its utility.
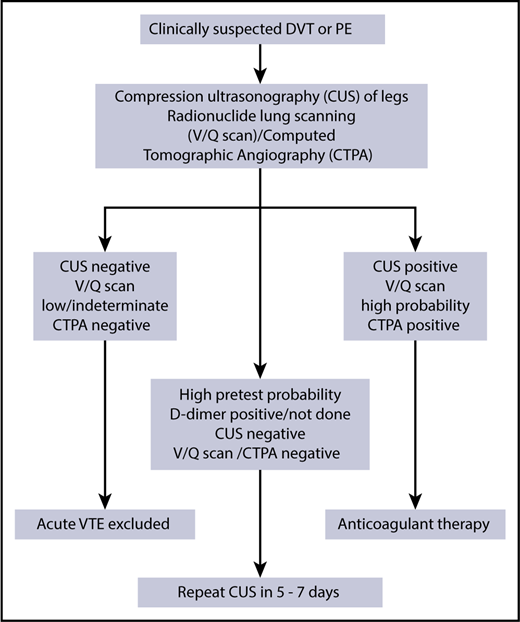
Our initial imaging includes the use of compression ultrasound Doppler for those suspected of upper- or lower-extremity DVT. We are not aware of any evidence that interpretation of compression ultrasound Doppler should be any different for a patient with SCD.
Multidetector computerized tomographic pulmonary angiography (CTPA) is currently a widely used test to detect PE. However, results may be confounded in patients with SCD experiencing ACS due to the high prevalence of in situ pulmonary thrombosis (17% [95% CI, 10%-23%]).48 Even in the absence of ACS, there is the possibility that subsegmental (or smaller) filling defects on CTPA may represent in situ sickling rather than a classic fibrin-rich clot. A further concern is the occurrence of contrast-induced acute kidney injury with CTPA, although we have not observed this with the newer nonionic low-osmolality contrast agents.
Radionuclide scanning (ventilation-perfusion [V/Q] scan) offers practical advantages over CTPA for the diagnosis of PE in SCD specifically by minimizing radiation exposure, absence of kidney injury, and well-defined diagnostic criteria. In one of our experiences (A.S.S.), diagnostic testing with V/Q scanning offers advantages over CTPA in patients who undergo frequent testing and offers specific advantages in establishing the diagnosis of chronic thromboembolic pulmonary hypertension.54 As with other populations, V/Q scanning is less useful in those individuals exhibiting pulmonary parenchymal abnormalities on plain chest radiographs.
Treatment of VTE in SCD
Treatment of acute VTE calls for urgent anticoagulation therapy to prevent extension, potentially fatal PE, and early recurrence of the thrombotic process, with no less than 3 months of anticoagulation because shorter treatment periods are associated with higher risk of recurrence.13 To date, the treatment of VTE in SCD relies on clinical guidelines established for VTE management in the general population because there are no clinical trials conducted to specifically inform anticoagulation practices in SCD. This includes consideration for thrombolytic therapy in appropriate situations. Currently, anticoagulation practice for the general population, as described in the American College of Chest Physicians (ACCP) 2016 guidelines,13 applies to individuals with SCD (summarized in Table 2). Nonetheless, there are special considerations for SCD patients that may modify the general recommendations, based on clinical judgement.
Summary of the approach to diagnosis and treatment of VTE in SCD
| VTE diagnosis and treatment approaches in SCD | |
|---|---|
| Diagnosis | • Compression ultrasonography (±Doppler) for deep venous thrombosis |
| • CTPA with nonionic low-osmolality contrast media | |
| o We do not routinely recommend red cell transfusion prior to contrast | |
| o Although less frequently performed V/Q scanning has clinical utility, especially when tested serially | |
| • D-dimer is routinely elevated in SCD precluding the high negative predictive value advantage this biomarker has in other settings | |
| Treatment | • Treatment as per ACCP 2016 guidelines with full-dose anticoagulation |
| o Potential for increased risk of bleeding in patients with MRA evidence for Moya Moya syndrome | |
| • Heparin, DOAC, or vitamin K antagonists are therapeutic options | |
| • In line with ACCP 2016 guidelines, our initial choice of anticoagulant is a DOAC if not contraindicated | |
| • Anticoagulate for at least 3 mo for VTE event | |
| • Consider extended anticoagulation in those with low bleeding risk even if the event was provoked by hospitalization for medical illness | |
| • Continue anticoagulation for catheter-associated upper-extremity thrombosis until catheter removal |
| VTE diagnosis and treatment approaches in SCD | |
|---|---|
| Diagnosis | • Compression ultrasonography (±Doppler) for deep venous thrombosis |
| • CTPA with nonionic low-osmolality contrast media | |
| o We do not routinely recommend red cell transfusion prior to contrast | |
| o Although less frequently performed V/Q scanning has clinical utility, especially when tested serially | |
| • D-dimer is routinely elevated in SCD precluding the high negative predictive value advantage this biomarker has in other settings | |
| Treatment | • Treatment as per ACCP 2016 guidelines with full-dose anticoagulation |
| o Potential for increased risk of bleeding in patients with MRA evidence for Moya Moya syndrome | |
| • Heparin, DOAC, or vitamin K antagonists are therapeutic options | |
| • In line with ACCP 2016 guidelines, our initial choice of anticoagulant is a DOAC if not contraindicated | |
| • Anticoagulate for at least 3 mo for VTE event | |
| • Consider extended anticoagulation in those with low bleeding risk even if the event was provoked by hospitalization for medical illness | |
| • Continue anticoagulation for catheter-associated upper-extremity thrombosis until catheter removal |
Adapted from Wun and Brunson.9
MRA, magnetic resonance angiography.
Type and intensity of anticoagulant therapy for VTE in SCD
As in the general population, there is no direct clinical evidence that the type of anticoagulant for the treatment of VTE should differ in patients with SCD. We typically treat SCD patients with acute VTE with direct oral anticoagulants (DOACs).13 However, estimation of glomerular filtration rate by serum creatinine can be inaccurate in patients with SCD potentially warranting the use of cystatin C, when available.55 Accurately estimating glomerular filtration rate and/or creatinine clearance guides appropriate selection of heparin and DOAC agents, all of which have varying degrees of renal clearance that can impact efficacy. For example, edoxaban may be less efficacious for nonvalvular atrial fibrillation when the creatinine clearance is >95 mL/min, which is not unusual in patients with SCD. Conversely, for varying creatinine clearance thresholds below 50 mL/min, dose reduction, and/or even use of alternative agents, is recommended for LMWH and DOACs.
For SCD patients with recurrent VTE, or bleeding on standard doses of nonwarfarin anticoagulants, we evaluate medication adherence, routinely check anti-Xa levels (for heparin), and consider testing DOAC drug levels. In SCD patients at high risk for bleeding, access to and availability of agents that can reverse the anticoagulant effect of DOACs may influence anticoagulant choice.56
Duration of anticoagulation
The duration of anticoagulation, beyond the minimum of 3 months for most patients, is determined by weighing the risk and seriousness of recurrent VTE with the risk of major bleeding. Data from the California cohort show the risk of recurrent VTE at 5 years to be nearly 37% in those averaging >3 admissions a year,12 and others have also reported a high rate of recurrent VTE.10 Interestingly, unpublished analysis of the California cohort reveals a high recurrence rate even in those with less severe disease (18% at 5 years, similar to men with unprovoked VTE13 ) with no difference whether the incident event was within or >90 days of a hospital admission. Therefore, a provoked VTE in a patient with SCD may be associated with a much higher risk of recurrence than a provoked VTE event in the general population.
The risk of major bleeding on therapeutic anticoagulation for VTE has typically been reported as low for patients without risk factors for bleeding.13 It is unknown whether SCD is associated with increased risk of bleeding on anticoagulation, absent other known risk factors, when compared with the general population of patients with VTE. However, preliminary data from the California cohort revealed a surprisingly high cumulative incidence of major bleeding of 2.9% at 6 months, and 5.0% at 1 year in SCD patients after incident VTE. Most of these episodes were gastrointestinal bleeding. This compares with 4% to 6% major bleeding incidence in patients treated for cancer-associated thrombosis with various anticoagulants on clinical trials57-59 and is higher than generally reported in large studies of non-SCD, noncancer patients, thereby stratifying SCD patients into the moderate to high risk for bleeding category.13 This information should also inform therapeutic considerations regarding the use of thrombolytic therapy for higher clot burden VTE in SCD patients.
The observation of a high risk of recurrent VTE, coupled with a relatively high risk of bleeding, is similar to the situation seen in patients with cancer.60-62 The current recommendation is to continue anticoagulation for cancer-associated thrombosis as long as there is active cancer.13,60 In the absence of clinical trials of anticoagulation in patients with SCD, we propose a conceptually similar approach. However, this must be carefully weighed against the increased risk of bleeding in these patients, which varies over time. This calculus would make one consider indefinite anticoagulation for a non–catheter-related VTE in a SCD patient regardless of whether the event was assessed to be provoked or not. Further work needs to be done to determine whether SCD patients with incident VTE can be risk stratified for recurrence, with duration of anticoagulation accordingly adjusted. Patient choice, health care costs associated with indefinite anticoagulation, and the potential long-term use of aspirin63 or lower doses of DOACs64,65 are also considerations that must be factored into making this decision.
For catheter-related VTE, we use the approach taken in other patients (Table 2). For symptomatic, upper-extremity catheter-related thrombosis, we recommend a minimum of 3 months of anticoagulation.66 If the catheter is functional and is required, we would continue therapeutic anticoagulation until catheter removal. Attention to the location of the catheter tip, and routine catheter care including anticoagulation flushes is also important to maintain catheter function and prevent mural thrombosis.
Cases revisited
Case 1
The patient’s symptoms resolved and after 3 months of treatment, he was reevaluated in clinic to discuss the risk and benefits of extending anticoagulation beyond 3 months to reduce his risk of recurrence. Despite a possible greater risk of recurrence with his family history, on the basis of severity of VTE (ie, DVT), and infrequent hospitalizations for SCD, he elected to discontinue anticoagulation after 3 months of therapy, due to bleeding risk-related concerns. Aspirin to reduce VTE recurrence, albeit less effectively than either warfarin or a DOAC but with a more favorable bleeding risk profile, was declined because he did not want an additional medication for an indefinite period.
Case 2
It was recommended to the patient that she start prophylactic-dose LMWH at the beginning of her second trimester through 6 weeks postpartum, with short interruption around the time of delivery. She did so, and had an uneventful spontaneous normal delivery. There was also discussion about the need for indefinite anticoagulation, given her prior VTE history and SCD. She decided against extended anticoagulation beyond the 6 weeks postpartum period, based on the provoked nature of her prior event, the time elapsed, her infrequent hospitalizations, and the increased risk for bleeding.
Case 3
The patient’s symptoms resolved with reduction in swelling and pain following the initiation of anticoagulation. The catheter position was confirmed to be appropriately located at the superior vena cava–right atrial junction. A V/Q scan was performed and ruled out the presence of PE. Due to concerns about the potential for dislodging a clot if erythrocytapheresis was performed, after 3 weeks of anticoagulation a repeat ultrasound was performed, which revealed organizing clot. Red cell exchange was then carried out without complication. Her anticoagulation will continue with a plan to stop therapy if the catheter is ever removed. An important area for future research is to prospectively assess the risk of catheter-associated thrombosis in SCD patients because it is known to be higher in patients with inherited thrombophilia.67
In conclusion, VTE is a frequent underrecognized clinical event that can complicate clinical manifestations adversely impacting the morbidity and mortality of SCD. The sickle genotypes HbSS and Sβ0-thalassemia, female sex, >3 hospitalization per year, functional hyposplenism, and presence of indwelling catheters are factors associated with increased risk. Restricting the use of central venous catheters and minimizing the exposure of patients to known risk factors for thrombosis could help prevent incident VTE. A high index of suspicion for VTE is required for early recognition and initiation of appropriate anticoagulation therapy. Diagnostic testing with compression ultrasonography for DVT, and V/Q scanning or CTPA for PE, is standard for objective confirmation of VTE in SCD. The diagnostic and prognostic value of D-dimer testing for VTE in SCD patients is yet to be clarified. Heparin, vitamin K antagonists, and DOACs are all effective agents in the treatment of VTE in SCD patients. A non–catheter-related VTE event in SCD justifies consideration of indefinite anticoagulation regardless of whether the event was provoked. However, the possible increased risk of bleeding in an SCD patient indicates that the choice of extended duration anticoagulation must be weighed carefully. Randomized clinical trials are warranted to identify optimal primary and secondary prevention strategies for VTE in SCD patients and to prospectively ascertain the risk or recurrence and bleeding.
Authorship
Contribution: A.S.S. and T.W. participated equally in the drafting and revision of the manuscript.
Conflict-of-interest disclosure: T.W. is a steering committee member and receives research funding from Janssen and Pfizer. A.S.S. declares no competing financial interests.
Correspondence: Arun S. Shet, Sickle Cell Branch, National Heart, Lung, and Blood Institute, National Institutes of Health, Building 10, Room 6S241 MSC 1589, 10 Center Dr, Bethesda, MD 20892-1589; e-mail: arun.shet@nih.gov.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal