

Abstract
Hemoglobinopathies are caused by genetic mutations that result in abnormal hemoglobin molecules, resulting in hemolytic anemia. Chronic complications involving the lung parenchyma, vasculature, and cardiac function in hemoglobinopathies result in impaired gas exchange, resulting in tissue hypoxia. Hypoxia is defined as the deficiency in the amount of oxygen reaching the tissues of the body and is prevalent in patients with hemoglobinopathies, and its cause is often multifactorial. Chronic hypoxia in hemoglobinopathies is often a sign of disease severity and is associated with increased morbidity and mortality. Therefore, a thorough understanding of the pathophysiology of hypoxia in these disease processes is important in order to appropriately treat the underlying cause and prevent complications. In this article, we discuss management of hypoxia based on three different cases: sickle cell disease, β-thalassemia, and hereditary spherocytosis. These cases are used to review the current understanding of the disease pathophysiology, demonstrate the importance of a thorough clinical history and physical examination, explore diagnostic pathways, and review the current management.
Introduction
Although the terms hypoxia and hypoxemia are often used interchangeably, they are not synonymous. Hypoxemia is defined as a condition where arterial oxygen tension (Pao2) is below normal. In young adults, the normal Pao2 ranges from 80 to 100 mm Hg (10.6-13.3 kPa) with an average of ∼95 mm Hg (12.6 kPa) and decreases with age with an average of ∼85 mm Hg (11.3 kPa) at 60 years. Hypoxia is defined as presence of low amounts of oxygen at the tissue level. Hypoxemia may lead to tissue hypoxia, but the Pao2 is only one factor in the delivery of oxygen to tissues. Additional factors include the oxygen affinity of the hemoglobin, the oxygen carrying capacity of blood, cardiac output, and blood flow distribution. The pH, temperature, carbon dioxide, and 2,3-DPG affect the oxygen dissociation curve and therefore affect oxygen delivery to the tissues. The presence of hemoglobin variants, the most common hemoglobinopathies being sickle cell disease (SCD) and the thalassemias, influence hemoglobin’s affinity for oxygen. Because of the defective hemoglobin molecule, anemia due to chronic hemolysis is the hallmark of severe hemoglobinopathies resulting in a reduced oxygen carrying capacity and a rightward shift of the dissociation curve.
There are 4 primary causes of hypoxia in both SCD and non-SCD patients: hypoventilation, diffusion impairment, cardiopulmonary shunt, and ventilation-perfusion inequality. Hypoventilation refers to a reduced amount of gas going to the alveoli per unit time. It is commonly caused by extrapulmonary diseases, and often the lung parenchyma is normal. In hypoxia caused by diffusion impairment, there is lack of equilibration between the partial pressure of oxygen in the pulmonary capillary blood and the alveolar gas. Examples include lung fibrosis that distorts the lung parenchyma and results in thickening of the alveolar walls and pulmonary hypertension (PH) that results in intimal wall thickening. Both mechanisms cause a barrier to efficient diffusion of oxygen by increasing the distance to diffusion of the oxygen molecule. A shunt allows mixing of deoxygenated blood that has not passed through the ventilated regions of the lung to mix with oxygenated blood, hence reducing the oxygen concentration. Intracardiac shunts are usually due cardiac malformations (eg, a ventricular septal defect), while extracardiac shunts may be anatomical, such as in pulmonary arteriovenous malformations, or due to intrapulmonary shunt referring to areas with decreased circulation due to vasoconstriction, resulting in ventilation-perfusion inequality. In general, ventilation-perfusion inequality is by far the most common mechanism of hypoxemia. It is caused by a mismatch of ventilation and blood circulation in various lung regions with the eventual mixture of oxygenated and deoxygenated blood arriving in the left ventricle. This is the case in conditions such as venous thromboembolism (VTE), acute chest syndrome (ACS), and atelectasis.
Chronic hypoxia in hemoglobinopathies may arise from one or a combination of these mechanisms. In SCD, for example, hypoxia has been associated with increased episodes of sickling resulting in painful crises, ACS, or development of PH. We speculate that in children, hypoventilation and ventilation-perfusion inequality are the predominant causes of hypoxia, with diffusion impairment becoming gradually important, but not the primary cause, with the development of fibrosis, restrictive physiology, and PH. Ultimately, patients with these complications have increased morbidity and mortality for any severity of disease. Timely recognition and management of hypoxia and its causes are therefore paramount to decreasing the associated sequela of chronic hypoxia. Here, we focus on the various mechanisms resulting in chronic hypoxia in hemoglobinopathies and hemolytic anemias and their treatment based on our current understanding of disease pathobiology.
Case 1: SCD
A 42-year-old African American male with HbSS is seen for an initial evaluation after moving from another state. He has frequent pain in his lower extremities and back. He has a history of left hip replacement due to avascular necrosis as well as cholecystectomy for gallstones. His medications include hydroxyurea in addition to daily analgesics. He reports no more than baseline lower back pain. He is found to have an oxygen saturation of 87% on room air.
An estimated 100 000 individuals or 1 out of every 400 African American newborns in the United States has SCD.1 Inherited in autosomal-recessive manner, a mutation in the sixth codon of the β-globin gene results in symptoms in the homozygous and compound heterozygous states. The resulting hemoglobin polymerize, resulting in formation of an intraerythrocyte viscous gel that induces deformed erythrocytes under hypoxic conditions. Within the vasculature, the resulting recurrent microvascular occlusion and hemolysis lead to tissue ischemia, causing pain episodes2 and ACS,3 and ultimately result in end-organ damage. Pain and ACS are the 2 most common morbidities, and ACS is a leading cause of mortality among individuals with SCD.4,5 Pulmonary complications of SCD include recurrent episodes of ACS leading to fibrosis of the lung parenchyma, VTE, and PH, and all of these can result in the development of chronic hypoxia.
Pulmonary complications are common and a major threat to the well-being of patients with SCD, accounting for a large proportion of deaths among these patients.5-8 According to the Cooperative Study of Sickle Cell Disease (CSSCD), in a prospective multicenter study of 3764 patients, >20% of adults likely had fatal pulmonary complications of SCD.5 Among the 299 patients enrolled in the long-term follow-up study of patients who participated in the Multicenter Study of Hydroxyurea in Sickle Cell Anemia, pulmonary disease was the most common cause of mortality, accounting for 28% of all deaths.9
A large number of patients with SCD demonstrate abnormal oxygen saturation at baseline with further desaturation during sleep and with exertion.10-12 In SCD, the oxyhemoglobin dissociation curve is shifted to the right, resulting in abnormally low arterial oxygen saturation even when oxygen partial pressure is normal. Although oxygen affinity in SCD patients is low under hypoxic conditions, it is normal at normal arterial oxygen saturation. Therefore, despite previous reports of unreliable oximetry in otherwise stable patients and underestimation of oxygenation in SCD patients presenting in painful crisis,13,14 a finding of abnormally low saturation on pulse oximetry readings in SCD constitutes reliable evidence of impaired gas exchange.15,16 Arterial blood gas measurements may confirm hypoxemia and help exclude abnormal hemoglobin variants whenever there is discrepancy between the oxygen saturation by pulse oximetry (Spo2) and arterial blood gas measurements (Sao2) as well as the Pao2.17
The etiology of hypoxia in SCD is likely multifactorial and often not fully understood (Table 1). Associated comorbidities include history and frequency of ACS and painful crisis, coexisting asthma, presence of PH and VTE and/or arterial thromboembolism, as well as SDB. Finally, the nonspecific term sickle cell chronic lung disease has been used to describe patients with lung disease associated with SCD. This wastebasket term is neither clinically helpful nor based on disease pathophysiology and should be abandoned from our lexicon. Specific diseases or physiologic patterns should be used to accurately describe lung disease in patients with SCD. It is with this framework in mind that a clinician should approach the diagnosis and treatment of chronic hypoxia in SCD.
Etiology of hypoxia in patients with SCD
| Mechanism of hypoxia | Causes in SCD |
|---|---|
| Hypoventilation | SDB (OSA, upper airway obstruction) Thoracic splinting due to chronic pain Restrictive pulmonary disease Reduced chest excursion due to hepatomegaly Central hypoventilation (eg, due to excessive use of narcotics) |
| Diffusion impairment | SCD-associated interstitial lung fibrosis PH Pulmonary vascular disease |
| Shunt | Intracardiac shunt (eg, ventriculoseptal defect) Extracardiac shunt Arterial-venous malformations Intrapulmonary shunt (eg, due to consolidation or atelectasis resulting in decreased perfusion to affected area) |
| Ventilation-perfusion inequality | Chronic VTE ACS Plastic bronchitis Obstructive lung disease without asthma Chronic airway inflammation due to asthma |
| Mechanism of hypoxia | Causes in SCD |
|---|---|
| Hypoventilation | SDB (OSA, upper airway obstruction) Thoracic splinting due to chronic pain Restrictive pulmonary disease Reduced chest excursion due to hepatomegaly Central hypoventilation (eg, due to excessive use of narcotics) |
| Diffusion impairment | SCD-associated interstitial lung fibrosis PH Pulmonary vascular disease |
| Shunt | Intracardiac shunt (eg, ventriculoseptal defect) Extracardiac shunt Arterial-venous malformations Intrapulmonary shunt (eg, due to consolidation or atelectasis resulting in decreased perfusion to affected area) |
| Ventilation-perfusion inequality | Chronic VTE ACS Plastic bronchitis Obstructive lung disease without asthma Chronic airway inflammation due to asthma |
OSA, obstructive sleep apnea; SDB, sleep-disordered breathing.
How do AHR and parenchymal lung disease contribute to hypoxia in SCD?
Up to 70% of pediatric patients with SCD have airway hyperreactivity (AHR).18-23 Data suggest that wheezing and obstructive lung disease may occur independently of asthma and are a marker of disease severity or a high output state.24,25 A diagnosis of asthma or recurrent episodes of wheezing in adults with SCD has been associated with increased SCD-related morbidity, including elevated rates of pain, likelihood of development of recurrent ACS,26 decreased lung function, and increased risk of death compared with adults without recurrent, severe wheezing.27-32 There is an association between elevated immunoglobulin E levels and asthma or airway hyperresponsiveness in individuals with and without SCD.33,34 Among other reasons, this association has been used to determine asthma as a separate comorbid condition in SCD.35 However, it is noteworthy to point out that additional, not yet clearly understood phenotypes of patients with wheezing in SCD exist. Pulmonary complications of SCD may mimic features of asthma such as wheezing, airway hyperresponsiveness, and obstructive lung disease,18,19,22,36 and many diagnoses of “asthma” in patients with SCD are likely related to SCD-specific mechanisms rather than traditionally defined asthma. This is exemplified by the high rates of positive response to methacholine as well as high rates of wheezing during ACS in patients without prior history of asthma. In a study of 1963 children and adults with SCD enrolled in the CSSCD, those with asthma and SCD died at younger ages, with a more than twofold increased risk of death compared with individuals with SCD without asthma.31 These data therefore indicate that asthma is linked to and increases the risk of acute vaso-occlusive episodes, ACS, and premature mortality.
Longitudinal studies suggest that ACS occurs in ∼50% of individuals with SCD over the course of follow-up.3 Repeated episodes of ACS have been postulated to result in development of fibrotic lung disease, which is a restrictive lung disorder evidenced by pulmonary function data and histologic findings of interstitial lung disease ILD and fibrosis37,38 (Figure 1). There are currently no guidelines for the diagnosis of restrictive lung disease in SCD, and epidemiological studies are lacking, but restrictive/fibrotic lung disease occurs most frequently in patients with multiple episodes of ACS and wheezing.39 Fibrotic lung disease can be detected by pulmonary function studies (PFTs) and high-resolution computed tomography of the chest. The most common noted abnormalities include reticular opacities, volume loss, and prominence of central vessels. This was also shown to correlate with lung function abnormalities.33 Taken together, a history of AHR or asthma contributes to the frequency of ACS, which may ultimately lead to development of conditions that are either individually or in combination associated with chronic hypoxia.
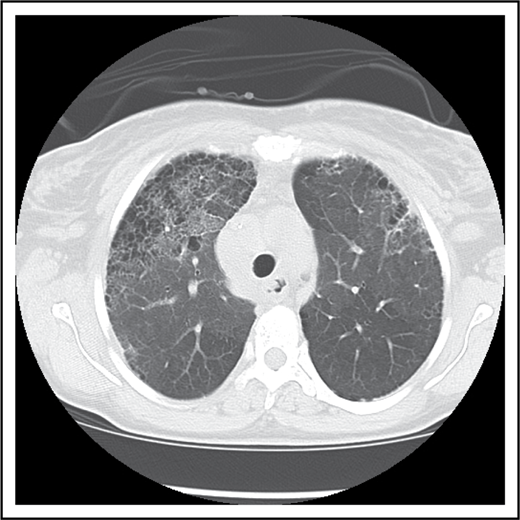
Pulmonary fibrosis complicating multiple episodes of ACS. Axial image of the chest computed tomography of a patient with SCD. Extensive fibrotic changes with honeycombing are noted anteriorly; reticular opacities are appreciated with an anterior predominance, and mild ground glass opacities are noted within the lungs.
Pulmonary fibrosis complicating multiple episodes of ACS. Axial image of the chest computed tomography of a patient with SCD. Extensive fibrotic changes with honeycombing are noted anteriorly; reticular opacities are appreciated with an anterior predominance, and mild ground glass opacities are noted within the lungs.
Given the high rates of AHR even without a diagnosis of asthma among patients with SCD, a thorough and detailed history of pulmonary symptoms, including cough and wheezing, should be obtained at each clinic visit. PFT testing with bronchodilator administration and, where appropriate, bronchoprovocation tests such as methacholine challenge may be helpful to aid in the diagnosis.
Additional history reveals that the patient has never been diagnosed with asthma but had episodes of wheezing during hospitalizations, with unclear response to a bronchodilator. On physical examination, he was found to have diminished breath sounds in the lung bases. Given this history of wheezing, PFTs were performed to evaluate for underlying AHR.
There is wide variability in PFT data in pediatric and adult patients with SCD. Both obstructive and restrictive physiology and decreased diffusion capacity of the lung for carbon monoxide (DLco) have been reported. Koumbourlis et al and Intzes et al in studies of pediatric patients both reported a 35% prevalence of airflow obstruction, with the remainder exhibiting a restrictive or normal PFT pattern.18,40 The majority of studies conducted in adult patients, however, have documented a predominant restrictive pattern.41-44 In the study by Klings et al, 310 (90%) patients with Hb-SS were found to have abnormal PFTs, with predominant restrictive physiology in 74% of their patients and independent decrease in DLco in some patients.45 Similar studies have also reported predominantly restrictive lung disease.46,47 The presence of restrictive physiology was associated with a trend toward more severe clinical disease, as exemplified by lower total hemoglobin and hematocrit concentrations, leukocytosis, and renal dysfunction.45 Additionally, Field et al studied the rate of decline in forced expiratory volume (FEV1) in adults in a longitudinal study and reported that this rate is increased among adults with SCD as compared with the general population for men and women (49 mL/year compared with 20-26 mL/year in the general population).48
Gas diffusion is also often impaired in SCD as demonstrated by DLco. An abnormal DLco is associated with abnormal oxygen saturation and is additionally associated with PH, thrombosis, hepatic and renal dysfunction which are indicative of severe vasculopathy45,49,50 as well as the presence of interstitial lung disease.51 Both dyspnea and exercise performance have also been linked to DLco parameters.52 Moreover, there is a gradual decrease in DLco associated with increasing age45 in keeping with worsening dyspnea with disease severity.
In summary, while children may have a mixed obstructive and restrictive lung physiology, lung disease in adult patients with SCD is predominantly characterized by mild restrictive physiology and may be associated with development of fibrotic lung disease and/or PH, both of which are associated with low baseline oxygen saturation. The presence of restrictive lung disease, however, is not synonymous with the presence of pulmonary fibrosis or PH, and severe fibrotic lung disease in adults with SCD is not common.
In 2014, the National Heart, Lung, and Blood Institute recommended against screening PFT in otherwise asymptomatic children and adults with SCD.53 However, when abnormal findings are obtained, studies should be repeated at a scheduled interval to ensure stability or normalization after an intervention. It is also recommended that the management of asthma in SCD be based on the current established National Asthma Education and Prevention Program guidelines, as there are no SCD-specific guidelines.54
The patient additionally reported frequent episodes of dyspnea with mild-to-moderate exertion.
While dyspnea is frequently reported in patients with SCD and is often nonspecific and causes are multifactorial, a consideration of PH as a cause for his abnormally low oxygen saturation should be explored. Presentation of PH is often nonspecific and similar to those caused by a known or unknown pulmonary or cardiac disease.55
Evaluation and treatment of PH
The prevalence of PH in SCD adults is ∼6% to 11% by right-sided cardiac catheterization,56,57 with an estimated 10% to 30% of HbSS and HbSC adults having a tricuspid regurgitant jet velocity (TRV) 2 standard deviations above the normal age-adjusted mean value.58,59 In young children and adolescents, an estimated 20% to 46% have an elevated estimated pulmonary artery systolic pressure39,60-63 screened by echocardiography and defined as a TRV ≥2.5 m/s. PH is associated with increased markers of hemolysis,60,62 a lower fetal hemoglobin level,64 a high reticulocyte count,61 and low oxygen saturation.63
In 2014, the American Thoracic Society published evidence-based consensus guidelines for the diagnosis and treatment of SCD-associated PH.59 Doppler echocardiography is recommended for the initial evaluation of patients with symptoms suggestive of PH (Figure 2). An elevated TRV on echocardiography is an indicator of possible PH, and it is a marker of early mortality in adults with SCD. The effects on survival rate in the pediatric SCD population are unclear.58 When echocardiography is unavailable or inadequate, NT-pro-BNP measurement can be a surrogate marker of presence of PH. The gold standard for the diagnosis of PH is right-sided cardiac catheterization, with PH being defined as a mean pulmonary arterial pressure ≥25 mm Hg at rest.59 An increased risk for mortality is defined as a TRV ≥2.5 m/s, an NT-pro-BNP level >160 pg/mL, or right heart catheterization (RHC)–confirmed PH. Additional evaluation with a 6MWD may be undertaken to measure functional capacity and to establish a baseline and track improvement with therapy.47,65 Finally, cardiac magnetic resonance imaging is an emerging diagnostic tool for myocardial dysfunction in patients with SCD.66
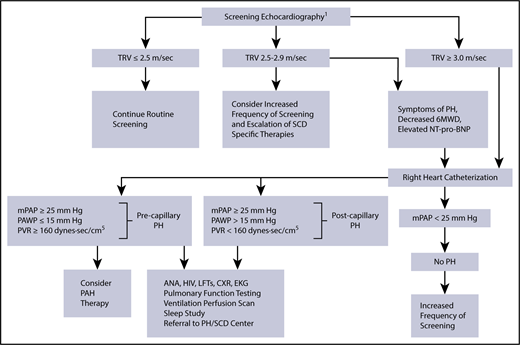
Proposed algorithm for evaluation of PH related to SCD. 6MWD, 6-minute walk distance; ANA, anti-nuclear antibody; CXR, chest radiograph; EKG, electrocardiogram; LFTs, liver function tests; mPAP, mean pulmonary artery pressure; NT-pro-BNP, N-terminal pro–brain natriuretic peptide; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance. 1The use of the term screening refers to mortality risk assessment. Echocardiography should be performed while patients are clinically stable. Patients with an mPAP between 20 and 25 mm Hg need further study, as they may be at increased mortality risk. PH therapy is to be considered on the basis of a weak recommendation and very-low-quality evidence. Reprinted from Klings et al,59 with permission.
Proposed algorithm for evaluation of PH related to SCD. 6MWD, 6-minute walk distance; ANA, anti-nuclear antibody; CXR, chest radiograph; EKG, electrocardiogram; LFTs, liver function tests; mPAP, mean pulmonary artery pressure; NT-pro-BNP, N-terminal pro–brain natriuretic peptide; PAWP, pulmonary artery wedge pressure; PVR, pulmonary vascular resistance. 1The use of the term screening refers to mortality risk assessment. Echocardiography should be performed while patients are clinically stable. Patients with an mPAP between 20 and 25 mm Hg need further study, as they may be at increased mortality risk. PH therapy is to be considered on the basis of a weak recommendation and very-low-quality evidence. Reprinted from Klings et al,59 with permission.
The use of hydroxyurea or chronic transfusion as well as targeted pulmonary arterial hypertension therapy is recommended as part of management therapy based on risk stratification and the etiology of PH, but data for randomized controlled studies are lacking.59 Supplemental oxygen should be administered to maintain arterial oxyhemoglobin saturation of at least 90% at rest or with exertion.67
VTE’s contribution to hypoxia in SCD
Although underrecognized, arterial and venous thrombosis with thromboembolism is prevalent in patients with SCD. SCD is associated with a hypercoagulable state due to endothelial dysfunction, chronic inflammation, and decreased levels of protein C, protein S, and anti-thrombin 3, as well as other coagulation factors, and heightened erythrocyte adhesion.68 Naik et al reported a prevalence of VTE of 25% in a cross-sectional study of 404 SCD patients in a single center with a median age at diagnosis of 30 years69 and a cumulative incidence of VTE of 11.3% by age 40 years in a separate multicenter retrospective study of patients enrolled in the CSSCD.70 Likewise, Brunson et al reported increased risk of VTE among women and those with severe disease. The cumulative incidence of VTE was 17.1% for severe SCD patients by age 40 years with a 36.8% 5-year recurrence rate.71 The largest study reporting a high prevalence of pulmonary embolism in patients with SCD was conducted using data from the National Hospital Discharge Survey. In this study, patients <40 years had a >3% higher risk of pulmonary embolism than African Americans without SCD.72 Pulmonary thrombi have also been noted in 17% of patients who underwent computed tomography scans during episodes of ACS.73 Further, up to 56% of patients with SCD had pulmonary artery thrombosis at autopsy.74-76
Venous thromboembolism is a known cause for hypoxia due to ventilation-perfusion inequality and can result in lung infarction. Chronic thromboembolism is also associated with development of PH in SCD. Anthi et al reported that patients with PH had significantly increased patchy pulmonary perfusion defects, with ventilation/perfusion scans of 3 patients suggestive of chronic thromboembolic pulmonary hypertension (CTEPH),47 but a diagnosis of thromboembolic PH is uncommon. Taken together, VTE is prevalent in patients with SCD and is a leading cause of morbidity and mortality. Further appropriate workup and management is based on established general population treatment guidelines,77 as SCD-specific guidelines are lacking.
SDB and nighttime hypoxemia in SCD
While SDB has been reported in up to 69% of pediatric patients with SCD,78 similar data are limited in adults. In small cohorts of adults followed prospectively, 44% to 50% of subjects were found to have SDB with mild to moderate OSA based on apnea-hypopnea index (AHI).79-81 The classification of OSA does differ between children and adults. In children, an AHI of 1 to 5 events per hour is classified as mild, 5 to 10 events per hour is moderate, and >10 events per hour constitutes severe OSA. In adults, mild OSA constitutes an AHI of 5 to 15 events per hour, moderate if 16 to 30 events per hour, or severe if >30 events per hour are recorded. Therefore, the clinical relevance of an AHI of 1 to 5 events per hour in adults is unclear and likely insignificant, even in SCD. This may also account for some of the variation in the prevalence of OSA between pediatric and adult studies. OSA is associated with poor sleep quality and daytime fatigue and without treatment has been associated with pulmonary and systemic hypertension, endothelial dysfunction, and stroke. It is unknown if these complications of OSA occur at an increased frequency in patients with SCD and OSA, but if left untreated, these complications would have serious implications in patients with SCD. Low waking or daytime pulse oximetry reading has been associated with nocturnal desaturation and OSA in children with SCD.82,83 In other studies, nocturnal hypoxemia has been detected even in the absence of OSA.81,84 Unlike adults, OSA in children with SCD is mostly associated with adenoid and tonsillar hypertrophy.78
Furthermore, the association between nocturnal hypoxia and clinical symptoms, including vaso-occlusive episodes resulting in pain, rates of ACS,85 and enuresis, and risk of neurologic complications in children with SCD has been documented, albeit in small nonprospective studies.10 However, in a recent prospective study of 140 children with SCD followed for a median of 4.9 years, low nocturnal oxygen saturation, higher obstructive AHI, and higher oxygen desaturation index were not associated with increased incidence of acute severe pain episodes.86 While the relationship between severity of OSA and related complications has been reported in small retrospective studies, this association is less robust, especially in adult subjects with SCD, because of the paucity of literature on the subject as well as conflicting findings, especially in regards to the incidence of pain episodes. Additional studies are needed to better define this association.
Given these data, a thorough history should be obtained to screen for symptoms suggestive of SDB. Such history may include (but should not be limited to) habitual snoring with gasping, daytime somnolence, and presence of tonsillar and adenoidal hypertrophy on examination. An overnight polysomnogram should be obtained to evaluate the presence and severity of OSA as well as the degree of nocturnal hypoxemia and hypercapnia in the appropriate clinical setting. Treatment should be tailored to the abnormalities identified during the workup (eg, tonsillectomy, continuous positive airway pressure, or supplemental oxygen).
On further questioning, the patient reported progressively worsening dyspnea over the previous 3 years. He had suffered 5 episodes of ACS and required mechanical ventilation and transfusion for 2 of these episodes. He underwent PFTs, including spirometry, that were suggestive of a moderate restrictive defect with a forced vital capacity of 68% predicted and FEV1 of 60% predicted with a ratio of 89. Restrictive lung physiology was confirmed on plethysmography with a total lung capacity of 63% predicted. His DLco was decreased at 52% predicted after correction for hemoglobin level. An arterial blood gas showed a Pao2 of 52 mm Hg (6.92 kPa) at rest. An echocardiogram was obtained and suggested absence of PH with a TRV jet velocity of 2.4 m/s and a NT-pro-BNP level of 130 pg/mL. A 6MWD put him at a functional capacity of 55% of predicted with desaturation to 76% at the completion of the test. Computed tomography of the chest demonstrated extensive areas of fibrosis (Figure 2).
Our patient was therefore diagnosed with fibrotic lung disease. Given the lack of interventions to reverse this condition, counseling was performed regarding the extent of his lung disease and the need for adherence to strategies to potentially slow or halt the progressive worsening of his lung disease. These included adjustment of his hydroxyurea to the maximum tolerated dose and more frequent follow-up visits to respond to any acute changes in his clinical health. He was placed on 1 L supplemental resting oxygen, which normalized his oxygen saturation at rest with titration to up to 3 L with ambulation. Figure 3 shows a proposed diagnostic algorithm for patients presenting with hypoxemia.
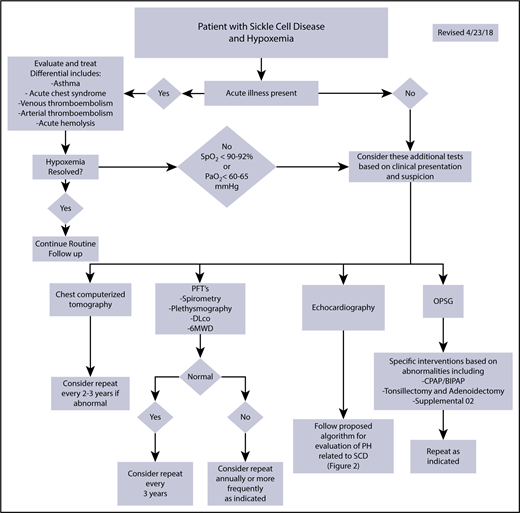
Proposed diagnostic workup algorithm for hypoxemia in patients with SCD. Proposed algorithm for evaluation of hypoxemia in patients with SCD and other hemoglobinopathies. BIPAP, bilevel positive airway pressure; CPAP, continuous positive airway pressure; OPSG, overnight polysomnogram. Echocardiography should be performed while the patient is clinically stable. All or some of the proposed studies may be obtained depending on the patient presentation and clinical suspicion for the underlying cause of hypoxemia.
Proposed diagnostic workup algorithm for hypoxemia in patients with SCD. Proposed algorithm for evaluation of hypoxemia in patients with SCD and other hemoglobinopathies. BIPAP, bilevel positive airway pressure; CPAP, continuous positive airway pressure; OPSG, overnight polysomnogram. Echocardiography should be performed while the patient is clinically stable. All or some of the proposed studies may be obtained depending on the patient presentation and clinical suspicion for the underlying cause of hypoxemia.
Case 2: BT
A 29-year-old female with transfusion-dependent β-thalassemia (BT) major presents for a scheduled blood transfusion. She is on deferoxamine to control her chronic iron overload. Other comorbidities include liver cirrhosis and insulin-dependent diabetes. Her surgical history includes splenectomy and cholecystectomy. She has no acute complains, but upon examination, she is found to have mild tachycardia and an oxygen saturation of 89% by pulse oximetry. Upon further questioning, she reports chronic dyspnea on moderate exertion. A diagnostic workup is initiated.
BTs are a congenital hemolytic anemias characterized by deficiency in the production of β-globin chains. They are caused by point mutations or, more rarely, deletions in the β globin gene on chromosome 11, leading to reduced (β+) or absent (β0) synthesis of the β chains of hemoglobin.87 Three forms exist: thalassemia minor, thalassemia intermedia (TI), and thalassemia major (TM). Severe anemia requiring regular red blood cell (RBC) transfusions is the hallmark of TM. These individuals typically present within the first 2 years of life. Patients with TI present later with moderate anemia and often do not require regular transfusions. Thalassemia minor is clinically asymptomatic, but some subjects may have moderate anemia.
Complications related to BTs are due to chronic hemolytic anemia, if not corrected, and include poor growth, jaundice from hemolysis, hepatosplenomegaly, and extramedullary hematopoiesis. Regular transfusions lead to iron overload, which may result in endocrine disorders, cardiomyopathy, and cirrhosis, among other complications. Treatment of BT includes regular RBC transfusions, iron chelation, and management of secondary complications of iron overload. In some circumstances, splenectomy is indicated. Several of the clinical complications related to thalassemia are related to complications of therapy such as transfusion-related iron overload, infection, thrombosis, possibly acute deterioration in pulmonary function associated with the use of the iron chelator deferoxamine, and splenectomy. In patients who are poorly transfused, extramedullary erythropoiesis can rarely cause pulmonary parenchymal masses and pleural effusions.
Several physiologic abnormalities arise in patients with BT, including restrictive and obstructive defects, hyperinflation, abnormal diffusion, and decreases in aerobic exercise capacity.88-90 There is little understanding of the pathophysiology of PFT abnormalities in this group of patients. Proposed mechanisms have included iron overload and correlate with transfusions, although it has not been established that this results from iron deposition in the lungs. Restrictive physiology and a low DLco have been described as the predominant pattern.91,92 In a cross-sectional study of 49 patients with BT without respiratory symptoms, forced vital capacity and FEV1 were decreased in 67% and 30% of individuals, respectively. Furthermore, these values were decreased in subjects with high ferritin (>2500 ng/dL) compared with those with low levels.93 Other studies have also reported an obstructive pattern in spirometry.94,95 Additionally, DLco is reduced in up to 62% of patients with BT.96-98 This is reportedly more frequent in patients with restrictive physiology.92 Increasing age is directly correlated with worse PFT.92,97 In spite of the varying PFT derangements, most patients remain clinically asymptomatic with abnormalities often detected during routine screening. Few studies have evaluated SDB in patients with BT. However, nocturnal desaturation is reported as a common phenomenon, especially in children with severe thalassemia.99
PH is a common complication in BT and a leading cause of mortality.100 The etiology of PH is multifactorial, and a significant proportion of patients develop PH due to left heart disease associated with iron overload (postcapillary PH). Echocardiographic screening studies suggest that ∼40% to 50% of patients with TI101 and 10% to 75% of patients with TM have echocardiographic evidence of PH.102-104 The largest trial using the gold standard RHC to identify PH was done by Derchi and colleagues,105 and it provides a better understanding of the precise prevalence in both TI and TM (importantly, the study excluded patients with chronic restrictive lung disease, a left ventricular ejection fraction of <50%, and evidence of chronic thromboembolic PH). Of 1309 patients prospectively enrolled in this study, ∼25% of the subjects had TI, while the remainder had TM. The majority of patients (94%) were considered unlikely to have PH following echocardiography with findings of TRV <3.0 m/s or were excluded. Thirty-three subjects with TRVs >3.2 m/s underwent RHC. Of these 33 subjects, 27 had precapillary PH (11 TM subjects and 16 TI subjects) and 4 had postcapillary PH (1 TM subject and 3 TI subjects). While the strict cutoff for TRV and the lack of case estimates for the nonscreened cohort likely underestimates the true prevalence of PH in β-thalassemia, this study does suggest a higher prevalence in TI (4.8%) than TM (1.1%). In this cohort, independent risk factors for PH included increasing age and a history of splenectomy. Splenectomy is thought to contribute to hypercoagulability due to elevated levels of abnormal RBCs, increase in intravascular hemolysis, and thrombocytosis that subsequently increases the risk of VTE.106-108
In summary, a combination of interstitial lung fibrosis, possibly due to chronic iron deposition in the lungs, chronic anemia, PH, and VTE may collectively or individually cause hypoxia in adult patients with BT. Given no standardized diagnostic protocol for this group of patients, we propose a similar systematic approach to the workup and management of patients with hypoxemia as that applied to the SCD population (Figure 3).
At the completion of the workup and the exclusion of significant left ventricular disease, parenchymal lung disease, and CTEPH, the patient was found to have moderate precapillary PH. Given the presence of symptoms and the severity of the PH, a multidisciplinary discussion occurred between the patient’s hematologist and a PH specialist. Her transfusion and iron chelation regimens were intensified with the goal of achieving a higher hemoglobin level, and specific PH therapy with a pulmonary vasodilator was initiated.
Despite its potential complications, chronic transfusion therapy in severe thalassemia has changed the clinical course of the disease and is likely to have a favorable impact in individuals with PH and other pulmonary complications of the disease. Combined with transfusions, iron chelation therapy prevents iron accumulation and the resulting oxidative tissue damage. This idea is supported by a report that in well-transfused, iron-chelated patients with TM, PH was completely prevented.109 Hydroxyurea modifies defective hemoglobin synthesis and reduces thrombocytosis, presumably decreasing hemolysis and hypercoagulability. In a recently published study of 584 patients with TI, use of these modalities seemed protective against development of PH.110 However, the study was not randomized, was retrospective, and used echocardiography for the diagnosis of PH. Other small studies and case reports in these patients have reported similar observations.111,112 Treatment of thalassemia patients with confirmed pulmonary arterial hypertension appears reasonable according to current PH treatment guidelines, but that has never been tested in a randomized trial. A favorable response to sildenafil has been reported in patients with TM and TI.113,114 In a recent open-label study of 10 patients with BT and an TRV >2.5 m/s on Doppler echocardiography, treatment with sildenafil resulted in a significant decrease in TRV and improved left ventricular end systolic/diastolic volume but did not change the 6MWD.115 There are also case reports of favorable response to bosentan116 and epoprostenol117 in patients with RHC-confirmed precapillary PH.
Case 3: inherited hemolytic anemia and HS
A 52-year-old female with hereditary spherocytosis (HS) is seen for consultation in the multidisciplinary PH clinic for an elevated TRV of 3.6 m/s on Doppler echocardiography. Her medical and surgical history includes splenectomy and regular scheduled transfusions. She is on deferoxamine for iron overload. She denies any current symptoms more than her baseline of shortness of breath with moderate exertion and chest tightness. Her oxygen saturation on pulse oximetry is 86%. Additional workup is initiated.
HS is the most common hemolytic anemia due to a red cell membrane defect with an incidence of ∼300 per million in northern European countries. It is inherited in an autosomal-dominant or recessive fashion with an additional inheritance mechanism of de novo mutations.118 HS is characterized by formation of spherical red cells with decreased deformability and increased membrane fragility that ultimately leads to hemolysis.
The incidence, prevalence, and mechanisms of chronic hypoxia in HS are poorly understood and understudied. Acute hypoxia is often a result of acute hemolysis with severe anemia resulting in reduced oxygen content in blood. There are no published studies examining PFTs or SDB in this group of patients. Similarly, the long-term complications related to hypoxia are unknown. One potential mechanism of chronic hypoxia in HS is through the development of PH.119-121 In these patients, splenectomy has been suggested as a factor for increased risk of development of arterial and venous thromboembolic events contributing to the development of CTEPH.119-121 The increased risk of thromboembolic events is attributed to thrombocytosis and increased release of prothrombotic factors.119 In small studies or case reports, PH has been documented by echocardiography and NT-pro-BNP, but confirmation with RHC is lacking.122
Further workup revealed the presence of severe precapillary PH. A ventilation perfusion scan demonstrated the presence of multiple segmental and subsegmental unmatched perfusion defects suggesting the presence of CTEPH. After multidisciplinary discussions with hematologists, radiologists, PH specialists, and thoracic surgeons, a decision to perform pulmonary endarterectomy at an experienced center was made.
The optimal management strategy for CTEPH is pulmonary endarterectomy.123 In patients who are not surgical candidates or who have residual postoperative PH, balloon pulmonary angioplasty and medical therapy with pulmonary vasodilators are alternative options.124 Data from a cohort of 19 patients with hemoglobinopathy or congenital hemolytic anemia (including 2 patients with HS) suggest that pulmonary endarterectomy is safe and efficacious in this group of patients. For patients with HbS exchange, blood transfusion was performed to reduce HbS to ≤20% for safe pulmonary endarterectomy with deep hypothermic circulatory arrest.125
Summary
The unifying pathobiological features of the 3 cases are hemolytic anemia and thrombophilia as well as its downstream consequences, all of which may individually or collectively contribute to the different mechanisms that result in hypoxia. As such, chronic hypoxia is prevalent in adults with hemoglobinopathies and hemolytic disorders, and its recognition and management are paramount in preventing potential chronic complications. Given the multiple potential etiologies of hypoxia in this patient population, a focused and pathophysiology-based diagnostic approach should be undertaken. More importantly, hypoxia should be also viewed as one of the manifestations of specific processes that should be individually addressed in addition to direct measures such as oxygen therapy given to correct oxygen desaturation. Unfortunately, most of the evidence used to base treatment recommendations in this patient population is borrowed from studies involving individuals without hemoglobin disorders. Large and well-designed management trials in these rare disorders are clearly needed.
Acknowledgments
This work was supported by the National Institutes of Health, National Heart, Lung, and Blood Institute (5R01HL111656).
Authorship
Contribution: E.M.M. and R.F.M. wrote the manuscript.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Roberto F. Machado, Division of Pulmonary, Critical Care, Sleep, and Occupational Medicine, Indiana University Department of Medicine, 980 W Walnut St, Walther Hall, R3 Room C400, Indianapolis, IN 46202; e-mail: robmacha@iu.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal