

Key Points
EPCR-PAR2 signaling regulates myeloid cell TLR4 responses independent of coagulation.
Abstract
Infection and inflammation are invariably associated with activation of the blood coagulation mechanism, secondary to the inflammation-induced expression of the coagulation initiator tissue factor (TF) on innate immune cells. By investigating the role of cell-surface receptors for coagulation factors in mouse endotoxemia, we found that the protein C receptor (ProcR; EPCR) was required for the normal in vivo and in vitro induction of lipopolysaccharide (LPS)-regulated gene expression. In cultured bone marrow–derived myeloid cells and in monocytic RAW264.7 cells, the LPS-induced expression of functionally active TF, assembly of the ternary TF-VIIa-Xa initiation complex of blood coagulation, and the EPCR-dependent activation of protease-activated receptor 2 (PAR2) by the ternary TF-VIIa-Xa complex were required for the normal LPS induction of messenger RNAs encoding the TLR3/4 signaling adaptor protein Pellino-1 and the transcription factor interferon regulatory factor 8. In response to in vivo challenge with LPS, mice lacking EPCR or PAR2 failed to fully initiate an interferon-regulated gene expression program that included the Irf8 target genes Lif, Iigp1, Gbp2, Gbp3, and Gbp6. The inflammation-induced expression of TF and crosstalk with EPCR, PAR2, and TLR4 therefore appear necessary for the normal evolution of interferon-regulated host responses.
Introduction
Blood clotting in response to vascular trauma is initiated by the contact of tissue factor (TF) with blood coagulation factors VII (FVII) and X (FX). Under normal circumstances, FVII and FX circulate in blood, whereas TF expression is largely confined to cells that are not in direct contact with blood. Injury to a blood vessel disrupts this tissue–blood barrier and results in the assembly of the initiation complex of blood coagulation, the ternary complex of TF with activated coagulation FX and FVII (TF-VIIa-Xa). Inflammation triggers TF expression in cells with direct exposure to blood, including the vascular endothelium and innate immune cells.1 This mechanistic coupling of TF-mediated coagulation activation to the inflammatory response to infection has been rationalized by considering the coagulation reaction as a specialized arm of the host defense that aids in the trapping and elimination of pathogens at sites of injury through the thrombin-dependent generation of fibrin. On the other hand, pro-inflammatory cell signaling by TF and the downstream coagulation proteases fXa and thrombin and systemic blood coagulation activation increase the pathologic burden of the host response by causing vascular thrombosis, resulting in compromised tissue perfusion and, in extreme cases, lethal disseminated consumptive coagulopathy.
The endothelial cell protein C receptor (EPCR; ProcR) supports the anticoagulant protein C (aPC) pathway function by augmenting the rate of protein C activation by the thrombin-thrombomodulin complex, and is also necessary for aPC-mediated anti-inflammatory signaling via protease-activated receptors (PARs).2,3 An erosion of the PC pathway function secondary to blood vessel injury, cytokine-mediated downregulation of EPCR and thrombomodulin, and proteolytic shedding of these receptors by neutrophil proteases is thought to contribute to the development of the severe procoagulant and proinflammatory state associated with bacterial sepsis and endotoxemia.4,5 Experiments in mice showed that EPCR deficiency in nonhematopoietic tissues (ie, endothelium) indeed exacerbated endotoxin-triggered systemic inflammation and coagulation activation, whereas the absence of EPCR from innate immune cells (ie, neutrophils, monocytes, and dendritic cells) had little, if any, effect.6 On the other hand, EPCR-dependent aPC signaling on innate immune cells appears necessary for mortality reduction by recombinant aPC in rodent models of sepsis and endotoxemia,7,8 possibly reflecting differences in the mechanisms of action of endogenous and therapeutically administered aPC. More recently, novel interactions of EPCR with the activated coagulation FXa and FVIIa, the αMβ2-proteinase 3 complex, the T-cell receptor complex on γδ T cells, and proteins derived from the malaria pathogen have been described that suggest additional functions of endothelial cell EPCR in the context of inflammatory or infectious disease (reviewed in Bouwens et al2 ). The importance of innate immune cell–associated EPCR in these contexts remains largely unknown.
Here, by examining how EPCR expression on mouse innate immune cells affects the endogenous host response to endotoxemia, we describe a novel biological function of EPCR as an essential coreceptor for TF-initiated cell-signaling events that are necessary for the normal evolution of TLR4- and interferon-regulated host defenses.
Materials and methods
Mice
C57Bl/6J mice were obtained from Jackson Laboratories (Bar Harbor, ME). Meox2Cre-EPCRloxP (“EPCR-null”), mice with reduced expression of TF (TFLOW), and PAR1-, 2- , 3-, or 4-deficient mice have been described.9-15 Mice expressing the R38E-PAR2 variant were generated via homologous recombination in C57Bl/6N embryonic stem cells (unpublished; Ruf et al, 2013). All other strains were backcrossed onto the C57Bl/6J background (≥12 generations). Animal experiments were in adherence with National Institutes of Health guidelines on the use of laboratory animals and approved by the Medical College of Wisconsin’s Institutional Animal Care and Use Committee.
Reagents
Lipopolysaccharide (LPS; Escherichia coli O55:B5) and hirudin were from Sigma (St Louis, MO). Anti-mouse TF antibody 21E10 was characterized previously.16 Active-site blocked FVIIai (Dansyl-Glu-Gly-Arg chloromethyl ketone-fVIIa), Nap5, and NapC2 were provided by Corvas International (San Diego, CA). Normal human pooled plasma was from Innovative Research (Novi, MI); calf thymus histone H3 was from Roche Diagnostics (Indianapolis, IN). Mouse TLR1-9 ligands (InvivoGen; San Diego, CA) were used at the following concentrations: Pam3CSK4: 200 ng/mL; heat-killed Listeria monocytogenes: 5 × 107 cells/mL; polyinosinic:polycytidylic acid: 2 μg/mL; LPS: 100 ng/mL; Flagellin Salmonella typhimurium: 2 μg/mL; FSL-1: 100 ng/mL; single-stranded RNA: 2 μg/mL; and ODN1826 CpG: 5 μM. Mouse anti-histone H4/3 BWA-317 was kindly provided by Dr Mark Monestier. Normal mouse plasma was obtained by puncture of the vena cava of heparin-anticoagulated wild-type mice (intraperitoneal infusion of unfractionated heparin 30 minutes before blood drawing), followed by centrifugation for 20 minutes/2000g at 4°C. Before use as culture supplement, plasma was incubated for 45 minutes at 56°C to minimize complement-mediated effects. SLIGRL PAR2 agonist peptide was purchased from Sigma-Aldrich (St Louis, MO). Antibodies used in flow cytometry experiments were purchased from Biolegend (Ter119, CD45.2 [clone 104], CD19 [clone 6D5], CD3ε [clone 145-2C11], CD11b [clone M1/70], Gr1 [clone RB6-8C5], CD31 [clone MEC13.3], Sca-1 [clone D7], c-kit [clone 2B8], CD150 [clone TC15-12F12.2]), eBioscience (Ly6C [clone HK1.4], CD274 [clone MINS], F4/80 [clone BM8], CD135 [clone A2F10], CD34 [clone RAM34], CD51 [clone RWV-7]), BD-Bioscience (Ly6G [clone 1A8]), StemCell Technologies (EPCR [clone RMEPCR1560]), and Santa Cruz (PAR2).
Endotoxemia and sepsis induction
LPS-endotoxemia and Staphylococcus aureus Newman (ATCC 25904, Manassas, VA) septic peritonitis were induced by intraperitoneal infusion of a predetermined median lethal dose of LPS (34 mg/kg) and bacteria (2 × 108 bacteria/mouse) causing approximately 50% lethality as previously described.8
TF activity
TF procoagulant activity was determined by a 2-stage chromogenic assay with Spectrozyme Xa substrate (Sekisui Diagnostics, Stamford, CT), recombinant mouse FVIIa (Novo Nordisk, Malov, Denmark), and human FX (Enzyme Research Laboratories, West Bend, IN) as described previously.18
Flow cytometry
Flow cytometry was performed on a BD LSRII or BD FACSAria III flow analyzer/sorter (BD Biosciences, San Jose, CA) and analyzed with BD FACS Diva or FlowJo software (TreeStar Inc, Ashland, OR).
Bone marrow transplants
Mice with hematopoietic TF deficiency were generated by transfer of 3 × 106 bone marrow cells isolated from the femur and tibia of TFLOW mice on a CD45.2 background into lethally irradiated (11 Gy) CD45.1 BoyJ recipients as described earlier.7 Mice with greater than 90% donor contribution (CD45.1/2 chimerism in peripheral blood cells) were used within 8 to 16 weeks after transplantation.
Tissue culture experiments
Mouse RAW264.7 cells (ATCC-TIB-71) were seeded at 0.75 × 106 cells/well into 6-well tissue culture grade dishes, grown overnight at 37°C with 5% CO2 to 70% to 80% confluence in Dulbecco’s modified Eagle medium (Lonza, Walkersville, MD) with 2% penicillin/streptomycin (Sigma), l-glutamine (4 mM, Sigma), and 10% (v/v) heat-inactivated fetal bovine serum (Atlanta Biologicals, Atlanta, GA). Cultures were rinsed with phosphate-buffered saline, and 3 mL of medium containing 10% v/v normal mouse or human plasma as a source of fX and fVII, and various reagents were added to each well. Mouse and human plasma yielded identical results, and human plasma was therefore used in the majority of experiments. All culture media contained 2 U of hirudin per milliliter to inhibit potential thrombin activity. After incubation, cell culture supernatants were aspirated and RNA was purified with TRIzol reagent (Invitrogen, Carlsbad, CA). Bone marrow–derived dendritic cells/macrophages (BMDCs) were prepared as described previously19 by 10-day culture of whole bone marrow in bacterial-grade dishes in the presence of recombinant mouse granulocyte macrophage colony-stimulating factor (GM-CSF, 20 ng/mL; R&D Systems, Minneapolis, MN). At day 10, cells were collected, suspended in R10 medium and seeded as described for RAW cell culture in tissue culture grade plastic dishes.
Gene expression analysis
Real-time reverse transcriptase–polymerase chain reaction (RT-PCR) was performed essentially as described previously7 using the Applied Biosystems 7500 Real-Time PCR System (Applied Biosystems, Foster City, CA), Quantum RNA 18S internal standards kit (used with competimers; Ambion, Foster City, CA), and PerfeCTa SYBR Green SuperMix (Quanta Biosciences, Gaithersburg, MD). Oligonucleotide primer sequences are listed in supplemental Table 3, available on the Blood Web site. Specificity of PCR was ascertained by melting curve analysis, agarose gel electrophoresis, and sequence verification of the amplicon. For array hybridization experiments, equal amounts of RNA from the indicated number of animals or tissue culture samples were combined before preparation of hybridization probes. Probe synthesis and hybridization to Affymetrix mouse genome 430 plus 2.0 arrays was conducted essentially as described previously.7 Image data were analyzed with Affymetrix GeneChip operating software and normalized with Robust Multichip Analysis (www.bioconductor.org/). The statistical significance of differential gene expression was derived through a Student t test and false discovery rates were determined with Significance Analysis of Microarrays software. Data were analyzed using Microsoft Excel and Ingenuity Pathways Analysis (Ingenuity Systems, Redwood City, CA).
Results
In vivo LPS responses are suppressed in mice with reduced EPCR expression
To characterize the effects of EPCR deficiency on the LPS response, we reexamined previously published gene expression data7 obtained by array hybridization analysis of spleen-resident CD11cPOS innate immune cells isolated from wild-type and LPS-treated mice with reduced EPCR expression (EPCRLOW) mice 16 hours after endotoxemia induction. Remarkably, diminished EPCR expression globally blunted the LPS response (Figure 1A) and was associated with a >1.5-fold difference in the hybridization signals of 15 564 (34%) probe sets. Inspection of expression data for 3040 genes induced >twofold by LPS and diminished >twofold in LPS-treated EPCRLOW mice (supplemental file 1) with the Upstream Regulator Tool of the Affymetrix Ingenuity Pathway Analysis (IPA) software indicated that reduced EPCR expression was associated with a marked suppression of the LPS response, interferon signaling, and Ticam1/TRIF-signaling downstream of TLR3 and TLR4 (Table 1). The differential expression of messenger RNA (mRNA) for the TLR3/4-TRIF–specific signaling adaptor protein Pellino-1 (Peli1), the interferon regulatory transcription factor 8 (Irf8; alias interferon consensus sequence binding protein, icsbp), the paracaspase Mucosa-associated lymphoid tissue lymphoma translocation protein 1 (Malt1), and the C-C motif chemokine 22 (Ccl22) were confirmed by quantitative RT-PCR analysis (Figure 1B). Reduced EPCR expression therefore appeared associated with a marked suppression of LPS- and interferon-regulated gene expression in innate immune cells.
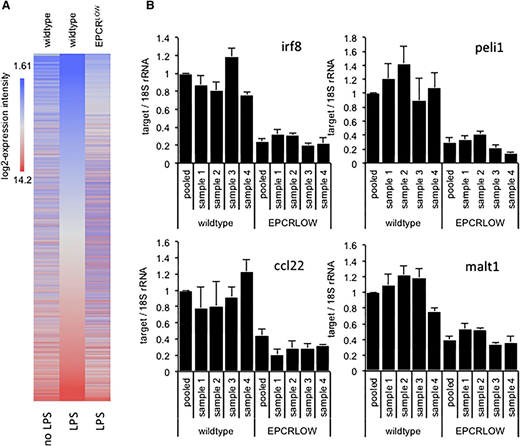
Diminished LPS responses in spleen CD11c cells isolated from EPCR-deficient mice. Total RNA was prepared from FACS-enriched spleen CD11c cells of unchallenged or LPS-treated wild-type mice and from EPCRLOW mice and analyzed by array hybridization of pooled RNA samples or by RT-PCR analysis of selected genes in samples from individual animals. (A) Heat map depicts the hybridization intensity of labeled complementary RNA prepared from pooled RNA samples to 44 105 gene probes of the Affymetrix mouse 430 2.0 genome array. Genes are arranged according to their expression level in LPS-treated wild-type mice (middle panel). EPCR deficiency was associated with a global suppression of the LPS response. (B) RT-PCR quantitation of Irf8, Peli1, Ccl22, and Malt1 mRNA relative to 18S ribosomal RNA (rRNA) in spleen CD11c cells of individual mice and in the pooled sample employed for array hybridization. Data represent the average ± standard deviation of 3 measurements in each sample, with the level in wild-type mice arbitrarily set to “1.”
Diminished LPS responses in spleen CD11c cells isolated from EPCR-deficient mice. Total RNA was prepared from FACS-enriched spleen CD11c cells of unchallenged or LPS-treated wild-type mice and from EPCRLOW mice and analyzed by array hybridization of pooled RNA samples or by RT-PCR analysis of selected genes in samples from individual animals. (A) Heat map depicts the hybridization intensity of labeled complementary RNA prepared from pooled RNA samples to 44 105 gene probes of the Affymetrix mouse 430 2.0 genome array. Genes are arranged according to their expression level in LPS-treated wild-type mice (middle panel). EPCR deficiency was associated with a global suppression of the LPS response. (B) RT-PCR quantitation of Irf8, Peli1, Ccl22, and Malt1 mRNA relative to 18S ribosomal RNA (rRNA) in spleen CD11c cells of individual mice and in the pooled sample employed for array hybridization. Data represent the average ± standard deviation of 3 measurements in each sample, with the level in wild-type mice arbitrarily set to “1.”
Effect of EPCR expression on candidate upstream effectors in CD11c spleen cells predicted by IPA analysis of 3038 genes differentially expressed in spleen CD11c cells of EPCRLOW mice
| Upstream regulator | Activation Z-score | Activation state | P value of overlap | Target molecule ratio |
|---|---|---|---|---|
| Interferon γ | 8.067 | Activated | 3.06 × 10−20 | 144/201 |
| LPS | 10.817 | Activated | 2.59 × 10−16 | 176/232 |
| TLR4 | 5.245 | Activated | 4.07 × 10−9 | 32/55 |
| TLR3 | 5.356 | Activated | 8.97 × 10−9 | 30/45 |
| IRF7 | 5.444 | Activated | 6.97 × 10−8 | 31/33 |
| Poly r(I:C) | 6.915 | Activated | 1.44 × 10−7 | 58/66 |
| IRF3 | 3.86 | Activated | 1.93 × 10−7 | 23/32 |
| TICAM1 | 4.98 | Activated | 6.22 × 10−5 | 26/26 |
| IRF8 | 2.98 | Activated | 4.91 × 10−4 | 16/28 |
| Upstream regulator | Activation Z-score | Activation state | P value of overlap | Target molecule ratio |
|---|---|---|---|---|
| Interferon γ | 8.067 | Activated | 3.06 × 10−20 | 144/201 |
| LPS | 10.817 | Activated | 2.59 × 10−16 | 176/232 |
| TLR4 | 5.245 | Activated | 4.07 × 10−9 | 32/55 |
| TLR3 | 5.356 | Activated | 8.97 × 10−9 | 30/45 |
| IRF7 | 5.444 | Activated | 6.97 × 10−8 | 31/33 |
| Poly r(I:C) | 6.915 | Activated | 1.44 × 10−7 | 58/66 |
| IRF3 | 3.86 | Activated | 1.93 × 10−7 | 23/32 |
| TICAM1 | 4.98 | Activated | 6.22 × 10−5 | 26/26 |
| IRF8 | 2.98 | Activated | 4.91 × 10−4 | 16/28 |
Candidate pathways altered by diminished EPCR expression were analyzed with the Upstream Regulator tool of the IPA software package. This tool employs statistical algorithms that measure how the observed changes in gene expression overlap with the experimentally observed effects of putative regulators, as curated from the literature. Pathways controlled by the indicated upstream regulators are predicted to be more highly activated in wild-type mice as compared with EPCR-deficient mice. The overlap P value measures enrichment of genes in the dataset that are known to be regulated by a regulator R without taking into account the direction of regulation. The activation Z-score predicts the potential activity of a given regulator by using experimental information about the direction of regulation of affected genes by the regulator. The target molecule ratio is the number of molecules in the tested dataset, divided by the total number of molecules in the Ingenuity Knowledge database that make up that pathway.
Poly r(I:C), polyriboinosinic:polycytidylic acid.
EPCR is required for normal in vitro LPS-induced gene expression
We next investigated the regulation of EPCR-dependent biomarkers (Irf8, Peli1, Malt1, and Ccl22) in GM-CSF–elicited BMDCs. Bone marrow cultures from wild-type and completely EPCR-deficient mice (ProcRLoxP-Meox2Cre) yielded comparable numbers of cells that were ∼85% homogeneous with respect to surface expression of CD11c, CD11b, and F4/80, with the remainder resembling F4/80POSCD11bLOW/INTCD11cNEG monocytic cells. Treatment of cultures with 100 ng/mL LPS for 3 hours resulted in the robust upregulation of all 4 biomarker mRNAs in wild-type BMDCs, but not in EPCR-deficient BMDCs (Figure 2A). In mouse myelo-monocytic RAW264.7 cells, biomarker mRNAs were induced by TLR4 ligands (LPS and histones), but not the TLR1/2 ligand Pam3CSK4, heat-killed L. monocytogenes (TLR2), high- or low-molecular-weight polyinosinic:polycytidylic acid (TLR3), the synthetic lipopeptide Pam2CGDPKHPKSF/FSL-1 (TLR2/6), single-stranded RNA (TLR7), or CpG (TLR9) (Figure 2B; Figure S2). The inflammatory in vitro response reflected in the induction of the EPCR-sensitive biomarkers in RAW cells therefore was specific to TLR4-ligands.

EPCR-dependent biomarker regulation in cultured cells. (A) GM-CSF–elicited BMDCs from wild-type or EPCR-deficient mice were treated for 3 hours with LPS (100 ng/mL) and mRNA levels of Irf8, Peli1, Ccl22, and Malt1 relative to 18S rRNA were measured by quantitative PCR. Data are expressed as the fold-difference relative to untreated wild-type cells and represent average ± standard deviation from 4 independent culture experiments. (B) RAW cells were treated for 3 hours with the indicated TLR ligands, and the abundance of Peli1 mRNA relative to 18S rRNA was measured by RT-PCR. Data represent the average ± standard deviation from 2 independent experiments with triplicate measurements of each sample. *P < .01 by pairwise comparison with levels in the untreated sample (Student 2-tailed t test). Ctrl, untreated sample; FLA-ST, Flagellin Salmonella typhimurium (TLR5 agonist; RAW cells lack TLR5); FSL-1: Pam2CGDPKHPKSF; HKLM, heat-killed L. monocytogenes. Corresponding data for Irf8, Ccl22, and Malt1 are shown in supplemental Figure 2.
EPCR-dependent biomarker regulation in cultured cells. (A) GM-CSF–elicited BMDCs from wild-type or EPCR-deficient mice were treated for 3 hours with LPS (100 ng/mL) and mRNA levels of Irf8, Peli1, Ccl22, and Malt1 relative to 18S rRNA were measured by quantitative PCR. Data are expressed as the fold-difference relative to untreated wild-type cells and represent average ± standard deviation from 4 independent culture experiments. (B) RAW cells were treated for 3 hours with the indicated TLR ligands, and the abundance of Peli1 mRNA relative to 18S rRNA was measured by RT-PCR. Data represent the average ± standard deviation from 2 independent experiments with triplicate measurements of each sample. *P < .01 by pairwise comparison with levels in the untreated sample (Student 2-tailed t test). Ctrl, untreated sample; FLA-ST, Flagellin Salmonella typhimurium (TLR5 agonist; RAW cells lack TLR5); FSL-1: Pam2CGDPKHPKSF; HKLM, heat-killed L. monocytogenes. Corresponding data for Irf8, Ccl22, and Malt1 are shown in supplemental Figure 2.
EPCR mediates PAR2 activation by the ternary TF-VIIa-Xa complex
Given that PAR2 activation amplifies TLR4 responses in immune and vascular cells,20,21 we investigated whether EPCR modulated the LPS response by enabling activation of PAR2 by the binary TF-VIIa or the ternary TF-VIIa-Xa initiation complexes of blood coagulation.22,23 In BMDCs prepared from mice lacking PAR1, PAR3, or PAR4, biomarker induction by LPS occurred normally, whereas PAR2 deficiency blunted Peli1 induction (Figure 3A; Figure S3). Over the course of the 3-hour exposure to LPS, BMDC and RAW cells exhibited a marked gain of procoagulant TF activity, measured via the activation of FX (Figure 3B). In wild-type BMDCs, LPS induction of all 4 biomarkers was suppressed by the function-blocking anti-mouse TF antibody 21E10,16 by active site–blocked FVIIai preventing formation of an active TF-VIIa-Xa complex, and by the specific FXa inhibitor nematode anticoagulant protein 5 (Nap5) (Figure 3C). Corresponding results were obtained with RAW cells (Figure 3D). The nematode anticoagulant protein C2 (NapC2), which inhibits TF-procoagulant activity, but stabilizes a conformation of the TF-VIIa-Xa complex that is conducive to PAR2 activation,22,23 had no effect on biomarker induction, ruling out that TF-procoagulant activity contributed to LPS-induced biomarker expression (Figure 3D). The mouse PAR2 agonist peptide SLIGRL rescued Peli1 induction by LPS in RAW cells in the presence of Nap5, or NapC2 and Nap5, but not inhibition by anti-TF antibodies or FVIIai. In BMDC isolated from a knock-in mouse strain expressing the R38E-PAR2 variant that is resistant to cleavage by all proteases, including FXa,24,25 biomarker induction by LPS was blunted relative to wild-type cells, but could be restored by costimulation with the PAR2 agonist peptide SLIGRL (Figure 3E). As in wild-type cells, SLIGRL-induced Peli1 expression was blocked by anti-TF-antibody. SLIGRL had no effect in BMDC lacking PAR2, documenting the specificity of this agonist response for PAR2. PAR2-dependent biomarker expression, whether induced by FXa or the PAR2 agonist peptide, therefore required association of PAR2 with an intact TF-VIIa-Xa complex. Together, these data showed that in vitro LPS-induced biomarker expression required the EPCR-dependent activation of PAR2 by FXa present in the ternary TF-VIIa-Xa complex.

Biomarker induction by LPS is due to PAR2 activation by the TF-VIIa-Xa complex. (A) Peli1 mRNA abundance relative to 18S rRNA in LPS-treated BMDCs prepared from wild-type (wt) mice and mice lacking the indicated PAR. Data are expressed as the fold increase over baseline levels in wild-type cells and represent the average ± standard deviation of 4 culture experiments with triplicate measurements per sample. *P < .01 for LPS-treated samples in comparison with wild-type cells via Student t test. Corresponding data for Irf8, Ccl22, and Malt1 are shown in supplemental Figure 3. (B) Tissue factor activity in BMDC and RAW cells was measured via the rate of FXa generation in a 2-stage amidolytic assay for FXa activity at baseline and after 3 hours of exposure to LPS. Data are expressed as the fold increase over baseline levels (no LPS) corrected for background activity (no FVII) and represent the average ± standard deviation of 6 independent experiments. (C-D) RAW cells or wild-type BMDCs were incubated for 3 hours with LPS (100 ng/mL), the indicated reagents (α-TF: anti-murine tissue factor antibody: 5 μg/mL; active-site blocked FVIIai, Nap5, NapC2: 500 nM; SLIGRL: 20 μM), and biomarker mRNA relative to 18S rRNA was quantitated by PCR. Data are expressed as the fold increase over no-LPS controls and represent the average ± standard deviation from 4 independent experiments. *P < .05 by comparison with cells treated with LPS alone (Student 2-tailed t test). (E) Peli1 mRNA regulation by LPS in BMDCs prepared from wild-type mice, a knock-in mouse strain expressing the cleavage resistant R38E-PAR2 variant, and PAR2-knockout mice (PAR2−/−). Data are the average ± standard deviation from 3 independent experiments. *P < .05 by Student 2-tailed t test as compared with LPS-treated wild-type cells.
Biomarker induction by LPS is due to PAR2 activation by the TF-VIIa-Xa complex. (A) Peli1 mRNA abundance relative to 18S rRNA in LPS-treated BMDCs prepared from wild-type (wt) mice and mice lacking the indicated PAR. Data are expressed as the fold increase over baseline levels in wild-type cells and represent the average ± standard deviation of 4 culture experiments with triplicate measurements per sample. *P < .01 for LPS-treated samples in comparison with wild-type cells via Student t test. Corresponding data for Irf8, Ccl22, and Malt1 are shown in supplemental Figure 3. (B) Tissue factor activity in BMDC and RAW cells was measured via the rate of FXa generation in a 2-stage amidolytic assay for FXa activity at baseline and after 3 hours of exposure to LPS. Data are expressed as the fold increase over baseline levels (no LPS) corrected for background activity (no FVII) and represent the average ± standard deviation of 6 independent experiments. (C-D) RAW cells or wild-type BMDCs were incubated for 3 hours with LPS (100 ng/mL), the indicated reagents (α-TF: anti-murine tissue factor antibody: 5 μg/mL; active-site blocked FVIIai, Nap5, NapC2: 500 nM; SLIGRL: 20 μM), and biomarker mRNA relative to 18S rRNA was quantitated by PCR. Data are expressed as the fold increase over no-LPS controls and represent the average ± standard deviation from 4 independent experiments. *P < .05 by comparison with cells treated with LPS alone (Student 2-tailed t test). (E) Peli1 mRNA regulation by LPS in BMDCs prepared from wild-type mice, a knock-in mouse strain expressing the cleavage resistant R38E-PAR2 variant, and PAR2-knockout mice (PAR2−/−). Data are the average ± standard deviation from 3 independent experiments. *P < .05 by Student 2-tailed t test as compared with LPS-treated wild-type cells.
TF, EPCR, and PAR2 are required for normal in vivo myeloid cell LPS responses
Within the bone marrow of unchallenged wild-type mice, EPCR expression was detected in immature macrophages (CD274-F4/80-CD34-CD135POS, CD31-Sca-1-c-kitHIGH, CD11b-Gr1-CD11cNEG). LPS challenge evoked the emergence of (EPCR-F4/80)POS cells that coexpressed Gr1 and CD11b (Figure 4A) and composed between 3% and 5% of the total Gr1CD11bPOS population. We therefore compared gene expression in Gr1CD11bPOS cells isolated from LPS-challenged TFLOW mice, EPCR-deficient mice, and PAR2-deficient mice. IPA analysis of 561 genes showing concordant regulation (induced >twofold by LPS in wild-type animals, diminished >twofold in all 3 LPS-challenged mutant mice; supplemental file 2) indicated that the loss/inhibition of receptor function impaired the activation of gene networks regulated by Ticam1/Trif signaling downstream of TLR3/4, and by interferon regulatory transcription factors (Table 2).

EPCR expression in bone marrow–resident macrophages. Flow cytometric characterization of bone marrow–resident EPCRPOS myeloid cells in unchallenged wild-type mice (no LPS) and 16 hours after LPS challenge (34 mg/kg). Scatterplots depict expression of EPCR, c-kit, and Sca-1 in CD45POS bone marrow cells. Histograms show surface expression of the indicated antigens (red line) in EPCRPOS cell populations in the indicated gates, with blue color indicating the signals obtained with isotype-matched nonimmune antibody. Cells coexpressing EPCR, Gr1, and CD11b express low levels of Sca-1 (as determined by back-gating; not shown).
EPCR expression in bone marrow–resident macrophages. Flow cytometric characterization of bone marrow–resident EPCRPOS myeloid cells in unchallenged wild-type mice (no LPS) and 16 hours after LPS challenge (34 mg/kg). Scatterplots depict expression of EPCR, c-kit, and Sca-1 in CD45POS bone marrow cells. Histograms show surface expression of the indicated antigens (red line) in EPCRPOS cell populations in the indicated gates, with blue color indicating the signals obtained with isotype-matched nonimmune antibody. Cells coexpressing EPCR, Gr1, and CD11b express low levels of Sca-1 (as determined by back-gating; not shown).
IPA analysis of 561 genes with concordant expression in in Gr1CD11bPOS cells isolated from mice with TF, EPCR, and PAR2 deficiency
| Upstream regulator | Activation Z-score | Activation state | P value of overlap | Target molecule ratio |
|---|---|---|---|---|
| TICAM1 | −3.36 | Inhibited | 4.97 × 10−9 | 14/15 |
| LPS | −5.28 | Inhibited | 1.60 × 10−8 | 44/57 |
| TLR4 | −2.86 | Inhibited | 3.69 × 10−8 | 12/20 |
| Interferon γ | −3.51 | Inhibited | 3.75 × 10−8 | 32/47 |
| MYD88 | −3.31 | Inhibited | 5.07 × 10−8 | 16/17 |
| TLR3 | −2.51 | Inhibited | 4.26 × 10−7 | 11/16 |
| Poly r(I:C) | −3.36 | Inhibited | 9.33 × 10−6 | 18/20 |
| IRF8 | −1.84 | Bias | 2.13 × 10−6 | — |
| IRF7 | −2.72 | Inhibited | 2.02 × 10−5 | 10 of 11 |
| IRF3 | −1.66 | Bias | 4.89 × 10−4 | — |
| Upstream regulator | Activation Z-score | Activation state | P value of overlap | Target molecule ratio |
|---|---|---|---|---|
| TICAM1 | −3.36 | Inhibited | 4.97 × 10−9 | 14/15 |
| LPS | −5.28 | Inhibited | 1.60 × 10−8 | 44/57 |
| TLR4 | −2.86 | Inhibited | 3.69 × 10−8 | 12/20 |
| Interferon γ | −3.51 | Inhibited | 3.75 × 10−8 | 32/47 |
| MYD88 | −3.31 | Inhibited | 5.07 × 10−8 | 16/17 |
| TLR3 | −2.51 | Inhibited | 4.26 × 10−7 | 11/16 |
| Poly r(I:C) | −3.36 | Inhibited | 9.33 × 10−6 | 18/20 |
| IRF8 | −1.84 | Bias | 2.13 × 10−6 | — |
| IRF7 | −2.72 | Inhibited | 2.02 × 10−5 | 10 of 11 |
| IRF3 | −1.66 | Bias | 4.89 × 10−4 | — |
Pathways controlled by the indicated upstream regulators are inhibited in mice with receptor deficiency.
EPCR and PAR2 deficiency attenuate LPS-induced in vivo expression of interferon-regulated genes
Irf8 is required for the normal inflammation-induced expression of a subset of interferon-responsive genes that includes leukemia inhibitory factor26 and a family of guanylate-binding proteins implicated in the host defense against diverse pathogens.27-29 Augmented expression of Irf8, Iigp1, Gbp2, Gbp3, and Gbp6 mRNA has also been described as part of a disseminated interferon-response gene expression signature of 84 genes that can be detected in immune cells and peripheral organs such as kidney and lung of mice after challenge with S. aureus enterotoxin,30 possibly reflecting immune cell infiltration of these tissues. We therefore analyzed RNA samples prepared 16 hours after endotoxemia induction by LPS from unfractionated lung and kidney tissue, and from fluorescence-activated cell sorter (FACS)-purified CD11bGr1POS cells. In the lung, Peli1, Irf8, Iigp1, and Lif were diminished in both EPCR- and PAR2-deficient mice as compared with wild-type animals (Figure 5A). In the kidney, the abundance of Peli1, Irf8, Lif, and Gbp2 was reduced to a similar extent in PAR2- and EPCR-deficient mice, whereas Iigp1 and Gbp6 were significantly reduced in EPCR-, but not in PAR2-deficient mice (Figure 5B). In bone marrow–resident Gr1CD11bPOS cells of mice lacking EPCR or PAR2, all of these candidate genes—Peli1, Irf8, Iigp1, Lif, Gbp2, Gbp3, and Gbp6—were markedly diminished (Figure 5C). A corresponding analysis of TFLOW mice yielded inconsistent results (Figure 5): although Irf8 was diminished in lung, kidney, and in BM-CD11bGr1 cells, other biomarkers showed either increased abundance (lung: Peli1, Lif, Gbp3; kidney: Peli1), diminished abundance (kidney: Iigp1, Gbp2, Gbp3, Gbp6; CD11bGr1-cells: Peli1, Lif, Gbp2), or unaltered expression (lung: Iigp1, Gbp2, Gbp3, Gbp6; CD11bGr1-cells: Iigp1, Gbp3, Gbp6). In summary, these data showed (1) that EPCR or PAR2 deficiency was indeed associated with defective in vivo expression of Irf8 target genes in bone marrow–resident Gr1CD11bPOS cells enriched in EPCR-expressing myeloid cells; (2) that even within the latter cells, partial reduction of TF expression reproduced the effect of EPCR or PAR2 deficiency only for some, but not all candidate genes; and (3) that EPCR, PAR2, or TF deficiency attenuated, but did not consistently abrogate the reported “interferon gene signature” in peripheral organs of mice exposed to S. aureus enterotoxin.

Attenuated interferon-regulated gene expression in the bone marrow and peripheral organs of LPS-challenged mice. Semiquantitative measurement of the indicated mRNAs relative to 18S rRNA by RT-PCR in (A) total lung, (B) kidney tissue, and (C) FACS-enriched bone marrow resident Gr1CD11bPOS cells of wild-type mice, mice lacking PAR2 or EPCR, and from TFLOW mice with reduced hematopoietic cell TF expression. Data are expressed as the fold increase relative to LPS-challenged wild-type mice and represent the average ± standard deviation from triplicate measurements of a single pooled sample generated by combining equal amounts of RNA prepared from 4 individual mice before reverse transcription (for lung and kidney tissue) or from 1 sample of RNA prepared from Gr1CD11bPOS cells isolated via FACS from the pooled bone marrows of 5 mice each. *P < .05 compared with wild-type controls.
Attenuated interferon-regulated gene expression in the bone marrow and peripheral organs of LPS-challenged mice. Semiquantitative measurement of the indicated mRNAs relative to 18S rRNA by RT-PCR in (A) total lung, (B) kidney tissue, and (C) FACS-enriched bone marrow resident Gr1CD11bPOS cells of wild-type mice, mice lacking PAR2 or EPCR, and from TFLOW mice with reduced hematopoietic cell TF expression. Data are expressed as the fold increase relative to LPS-challenged wild-type mice and represent the average ± standard deviation from triplicate measurements of a single pooled sample generated by combining equal amounts of RNA prepared from 4 individual mice before reverse transcription (for lung and kidney tissue) or from 1 sample of RNA prepared from Gr1CD11bPOS cells isolated via FACS from the pooled bone marrows of 5 mice each. *P < .05 compared with wild-type controls.
Discussion
The current work documents a previously unknown function of EPCR in innate immune cells that is necessary for the normal evolution of the host responses to endotoxemia. Our findings indicate that EPCR-dependent activation of PAR2 signaling amplifies a specific component of the gene expression program initiated in response to TLR4 engagement by pathogen-derived danger signals, such as LPS, and by tissue damage–associated signals, such as histones. The accessory function of PAR2 for a productive TLR4/interferon host response described here is consistent with earlier observations by others that crosstalk and physical interaction between PAR2 and TLR4 are necessary for the normal engagement of TRIF and MyD88-dependent TLR4 signaling pathways,21 that PAR2 interacts with TLR4 to regulate vascular reactivity in naive and endotoxin-challenged rodents,20 and with the enhancing effect of PAR2 on dendritic cell maturation.31 The proteases that activate PAR2 in the context of inflammation have not been identified in these latter reports. Our analyses of RAW cells and BMDCs show that under in vitro conditions the PAR2-activating protease is FXa, generated as a result of inflammation-induced expression of TF, and assembly of the ternary TF-VIIa-Xa complex. These findings extend earlier analyses of TF signaling in human and mouse cell models, in which EPCR selectively promoted PAR2 activation by the ternary TF complex, but not by the binary complex of TF-VIIa. A notable feature of in vitro TF-EPCR-PAR2 signaling in RAW cells as well as in BMDCs was that inhibition of ternary complex formation by TF-blocking antibodies or by active site blocked FVIIai abrogated the ability of the PAR2 peptide agonist SLIGRL to support normal inflammatory gene expression. This suggests that TF expression is not solely required for FX activation, but also modulates PAR2 responsiveness to nonproteolytic agonists, possibly by recruiting PAR2 into a signaling-competent membrane platform, by regulating the cell-surface localization, or endocytosis of PAR2, or a combination of such mechanisms.
Several lines of evidence support the notion that the EPCR-dependent activation of PAR2 by the TF-VIIa-Xa complex is a physiologically relevant modulator of the in vivo inflammatory response, and that the specific component of this response regulated by TF-EPCR-PAR2 signaling aligns with Ticam1/TRIF-mediated signaling pathways downstream of TLR4 and with myeloid cell functions regulated by interferon-regulated transcription factors. First, unbiased in silico pathway analyses of genome-wide differential gene expression in spleen CD11cPOS myeloid cells isolated from LPS-challenged mice identified these pathways as the most likely targets of EPCR-dependent signaling. Second, the biomarkers Irf8 and Peli1, which were extracted from these analyses as surrogate readouts for in vitro analyses of EPCR signaling merely because of their robust in vivo and in vitro differential gene expression, are known to regulate the TRIF-dependent arm of TLR4 signaling and interferon-mediated functions of myeloid cells. The ubiquitin E3 ligase Pellino-1 is an essential adaptor protein in the TRIF-mediated TLR3/4 signaling pathway.32 Peli1 deficiency renders mice resistant to LPS-induced septic shock and inhibits TLR4- and TLR3-triggered inflammatory cytokine production. The LPS-induced augmentation of Peli1 expression in BMDCs is itself dependent on an intact TRIF pathway,33 indicating that PAR2 signaling might amplify this feedback loop. Notably, the latter study also showed that TRIF deficiency markedly suppressed interferon-regulated host defense genes of the immunity-related guanosine triphosphatase and guanylate-binding protein families. Activation of the Stat1-interferon pathway via the TRIF-mediated arm of TLR4 signaling, and the ensuing Stat1-mediated expression of IRF family transcription factors, including Irf8, are necessary for the amplification of LPS responses and sustained interferon signaling.34-36 The transcription factor Irf8 further regulates the normal differentiation and inflammatory maturation of dendritic cells and macrophages.37,38 Third, to establish whether EPCR signaling altered the organismic response to endotoxin challenge, we measured in mice with genetically disrupted TF-EPCR-PAR2 function the expression of additional, downstream TRIF/interferon-regulated candidate genes in the kidney, lung, and bone marrow–resident Gr1CD11bPOS cells. In EPCR- and PAR2-deficient animals, some, but not all, candidate genes were attenuated in the lung or kidney. This differential effect of EPCR or PAR2 deficiency in different anatomical locales dovetails the reported time- and organ-selective extent of the interferon gene signature seen in enterotoxin-challenged mice and possibly reflects organ-specific differences in the extent and cell type selectivity of inflammatory cell infiltration, or partial compensation by locally produced cytokines. In line with this interpretation, all candidate genes were consistently suppressed in EPCR- and PAR2-deficient Gr1CD11bPOS cells that reside and undergo inflammatory maturation within the bone marrow. Interestingly, the expression of EPCR clearly identifies a specific subset of bone marrow Gr1CD11bPOS cells that bear a resemblance to the inflammation-induced population of so-called “myeloid-derived suppressor cells” (MDSCs). The latter are a heterogeneous population of myeloid cells with immunoregulatory functions in the context of sepsis, cancer, and trauma.39,40 Of note, MDSC differentiation is in part regulated by Irf841 and may be sensitive to the EPCR ligand aPC.42 It is currently unknown whether the EPCR-expressing Gr1CD11bPOS subset exerts similar, likely stage- and context-specific immunoregulatory functions as MDSC.
In contrast to the compelling in vitro evidence for the ternary TF complex being the relevant PAR2 activator, the analysis of mice expressing low levels of a human TF transgene yielded inconsistent data, showing for some markers increased, rather than the predicted decreased gene expression, or an absence of an effect. Still, the direct RT-PCR quantitation of marker expression and the pathway analysis of global gene expression in Gr1CD11bPOS cells (Table 2) support the working model that TF contributes to the overall in vivo inflammatory response regulated by EPCR-PAR2 signaling. The interpretation of findings in TFLOW mice is further confounded by the unknown effects of residual TF signaling, potential cross-species effects caused by expression of the human TF transgene in TFLOW mice, by the unknown in vivo consequences of reduced TF procoagulant activity for inflammatory gene expression, and by the potential impairment of myeloid cell migration mediated by alternatively spliced TF.43 We also note that others have reported a “paradoxically” increased granulocyte accumulation in the lung of mice with reduced hematopoietic TF expression44 (which would be consistent with the enhancing effect of reduced Irf8 expression on granulopoiesis38,45 ), whereas pharmacologic targeting of the ternary TF-VIIa-Xa complex with NapC2 (which blocks TF-initiated coagulation activation, but preserves TF-EPCR-PAR2 signaling, as shown in Figure 3C-D) had no such effects.46,47
The described contributions of TF, EPCR, and PAR2 to the normal integration of TLR4- and interferon-regulated gene expression extend the biological rationale for the mechanistic coupling of inflammation and TF-mediated coagulation activation. Traumatic tissue damage causes blood loss and at the same time disrupts the normal tissue barriers that protect against pathogen invasion. Preformed TF expressed in perivascular tissue initiates thrombin generation to provide immediate local hemostasis and may support fibrin-dependent mechanisms of bacterial entrapment and clearance by neutrophils at sites of injury. On the other hand, as shown here, the inflammation-induced de novo expression of TF on innate immune cells may also augment via EPCR- and PAR2-dependent crosstalk with TLR4 a gene expression program that is required for the normal acquisition of fully functional host defenses. The in vivo activation of this program likely occurs not only in response to pathogen-derived signals, but also can be triggered by endogenous TLR4 ligands released from damaged tissue, such as histones (as shown here), or the chromatin-associated high-mobility group box1 protein. Activation of this pathway may therefore be associated with the host response to a wide spectrum of pathogens as well as noninfectious inflammatory states. For example, Peli1 activation in microglial cells is a key modifier of neuroinflammation in experimental autoimmune encephalomyelitis models,48 and activation of the IRF8 gene signature is also associated with the neuroinflammatory sequelae of cerebral malaria.27 Notably, partial IRF8 deficiency impairs innate and adaptive immune defenses against the blood-stage malarial parasite Plasmodium chabaudi AS.49 EPCR has recently been identified as a receptor for variants of the malaria pathogen–derived protein PfEMP1 that are associated with severe disease. In contrast, PfEMP1 variants associated with less severe disease do not interact with EPCR.50 An interesting question raised by our findings is whether the binding of PfEMP1 to EPCR could disrupt EPCR-dependent PAR2 activation and thereby allow the parasite to subvert Irf8-dependent host defenses. Similarly, it is currently unknown to what extent the well-documented anti-inflammatory effects of the EPCR ligand aPC involve the competitive inhibition of EPCR-dependent PAR2 signaling on innate immune cells. The evolutionary conserved functions of Peli1, Irf8, and the Malt1-Bcl10-Card9 complex in humans, together with emerging evidence for the role of these pathways in human inflammatory disease,32,51-53 warrant further investigations into the potentially conserved function of the TF-EPCR-PAR2 signaling axis in human immune cells.
Supplemental material
Supplemental file 1 contains the set of EPCR-regulated genes in spleen CD11c cells that were analyzed with IPA software. Supplemental file 2 contains expression data from bone marrow–resident Gr1CD11bPOS cells and the subset of genes coregulated by EPCR, PAR2, and TF used for IPA analysis. Supplemental file 3 contains the primer sequences for mRNAs measured via RT-PCR. Supplemental Figure 2 provides supplemental data to Figure 2 for the biomarkers Ccl22, Malt1, and Irf8. Supplemental Figure 3 provides supplemental data to Figure 3A for the biomarkers Ccl22, Malt1, and Irf8.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
The authors thank Dr Nigel Mackman for providing bone marrow from TFLOW mice, Dr Mark Monestier for generously providing anti-histone H3/4 antibody BWA3, and Drs Charles and Naomi Esmon for providing MeoxCre-EPCRflox mice.
This work was supported by the National Institute of Health, the National Institute of Allergy and Infectious Disease (AI080557; H.W.), and the National Heart, Lung, and Blood Institute (HL44612-14; HL093388; HL77753; HL52246); Bridge Funding from the American Society of Hematology (H.W.); and the Ziegler Family Research Chair Foundation (H.W.).
Authorship
Contribution: H.P.H.L., E.J.K., I.H., and S.B. conducted experiments, analyzed data, prepared figures, and cowrote the manuscript; M.Z. and F.B. conducted experiments and prepared data; S.J. and M.J.H. analyzed gene expression experiments and cowrote the manuscript; J.H.G. and W.R. contributed to the experimental design and data interpretation, provided reagents, and cowrote the manuscript; and H.W. conducted experiments and analyzed data, cowrote the manuscript, and was responsible for overseeing and coordinating this work.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
The current affiliation for H.P.H.L. is Sutton Arthritis Research Laboratory, Royal North Shore Hospital, St Leonards, New South Wales, Australia.
Correspondence: Hartmut Weiler, Blood Research Institute, BloodCenter of Wisconsin, 8727 Watertown Plank Rd, Milwaukee, WI 53226; e-mail: hartmut.weiler@bcw.edu.

This feature is available to Subscribers Only
Sign In or Create an Account Close Modal