

Key Points
We used molecular dynamics, saturation mutagenesis, and enzymologic screening to develop a glutaminase-free mutant (Q59L) l-ASP.
We then used Q59L to show that glutaminase activity is not required for l-ASP activity against ASNS-negative cancer cells.
Abstract
l-Asparaginase (l-ASP) is a key component of therapy for acute lymphoblastic leukemia. Its mechanism of action, however, is still poorly understood, in part because of its dual asparaginase and glutaminase activities. Here, we show that l-ASP’s glutaminase activity is not always required for the enzyme’s anticancer effect. We first used molecular dynamics simulations of the clinically standard Escherichia colil-ASP to predict what mutated forms could be engineered to retain activity against asparagine but not glutamine. Dynamic mapping of enzyme substrate contacts identified Q59 as a promising mutagenesis target for that purpose. Saturation mutagenesis followed by enzymatic screening identified Q59L as a variant that retains asparaginase activity but shows undetectable glutaminase activity. Unlike wild-type l-ASP, Q59L is inactive against cancer cells that express measurable asparagine synthetase (ASNS). Q59L is potently active, however, against ASNS-negative cells. Those observations indicate that the glutaminase activity of l-ASP is necessary for anticancer activity against ASNS-positive cell types but not ASNS-negative cell types. Because the clinical toxicity of l-ASP is thought to stem from its glutaminase activity, these findings suggest the hypothesis that glutaminase-negative variants of l-ASP would provide larger therapeutic indices than wild-type l-ASP for ASNS-negative cancers.
Introduction
l-Asparaginase (l-ASP) is an enzyme drug used in combination with vincristine and a glucocorticoid (eg, dexamethasone) to treat acute lymphoblastic leukemia (ALL).1,2 We3-6 and others7 have reported a rationale for testing l-ASP against low-asparagine synthetase (ASNS) solid tumors as well. l-ASP’s primary known enzymatic activity is deamidation of asparagine to aspartic acid and ammonia, but it also deamidates glutamine to glutamic acid and ammonia, although with lower affinity and lower maximal rate. l-ASP therapy is often limited by toxic side effects that are generally attributed to the glutaminase activity.8,9 Those side effects often preclude completion of the full treatment regimen, resulting in poor outcome.10 The question that arises, however, is whether the therapeutic index of l-ASP could be increased by decreasing its glutaminase activity8,9 or whether that would also decrease the anticancer effect commensurately.
One side of the debate hypothesizes that l-ASP’s therapeutic index can be improved by increasing glutaminase activity. In support of that hypothesis, data collected over the last decade have suggested that glutaminase activity generally increases the efficacy of l-ASP and is sometimes required to achieve an anticancer effect. Those studies have reported asparaginase activity to be expendable.11-19 However, clinical evaluation of the high-glutaminase l-ASP from Acinetobacter glutaminasificans yielded considerable toxicity,20 suggesting that glutamine depletion increases toxicity to a greater extent than it increases anticancer activity, resulting in a low therapeutic index. Continued efforts to develop glutaminase-based therapies should clearly proceed with caution.
The other side of the debate hypothesizes that l-ASP’s therapeutic index can be improved by decreasing glutaminase activity. In a previous test of that strategy, a “glutaminase-free” l-ASP variant from Wolinella succinogenes was isolated, characterized,15 and evaluated clinically through a US National Cancer Institute Rapid Access to Intervention Development (NCI RAID) grant between 2001 and 2008. Unexpectedly, however, W succinogenesl-ASP was found to possess glutaminase activity and to be toxic in patients. The decision was made to not file an investigational new drug application. Because W succinogenesl-ASP was not truly “glutaminase-free,” the question whether reduced glutaminase activity improves therapeutic index remains unanswered.
Here, we report a new contribution that supports both sides of the debate. Because previous studies found W succinogenesl-ASP to be toxic, we first reasoned that a “glutaminase-free” l-ASP should be derived from an isoform that is safe and effective. Accordingly, our work focuses on the clinically used Escherichia colil-ASP. Molecular dynamics (MD) simulations of that enzyme guided rational engineering of a glutaminase-deficient variant. Residues that preferentially interacted with glutamine over asparagine but were not essential to the enzymatic conversion were chosen as candidates for saturation mutagenesis. The top candidate was amino acid Q59. We identified a mutant (Q59L) that exhibits at most a small percentage of the original glutaminase activity yet retains >60% of wild-type (WT) asparaginase activity. Finally, we found Q59L to exhibit selective anticancer activity against ASNS-negative leukemia cell lines, whereas the WT enzyme exhibited ASNS-independent toxicity.
Materials and methods
MD simulations
Details of the MD simulations and associated analyses are provided in the supplemental Methods (see supplemental Data available at the Blood Web site).
Compounds and plasmids
Elspar (Escherichia colil-Asp) was purchased from Lundbeck Pharmaceuticals. The gene coding for E colil-Asp II (ansB; referred to here as l-ASP) was polymerase chain reaction–amplified from genomic DNA of E coli top 10 strain (Invitrogen). To facilitate purification of recombinant proteins, a 6x histidine tag was incorporated in the forward primer (primers 97 and 98; all primer sequences are listed in supplemental Table 1).
Determination of asparaginase and glutaminase enzyme activity
Asparaginase enzyme activity was measured using an established colorimetric asparaginase assay. The assay, which uses l-aspartic acid β-hydroxamate as a substrate,21 was modified as described in supplemental Methods. Glutaminase enzyme activity was measured using a Glutamate Assay kit (Abcam) according to the manufacturer’s instructions (see supplemental Methods for details). Asparaginase and glutaminase activities were also determined using a highly sensitive liquid chromatography–mass spectrometry (LC-MS)/MS assay developed by our laboratory.22 Limits of detection for asparagine, aspartic acid, glutamine, and glutamic acid were 250, 150, 16, and 22 nM, respectively.
Mutagenesis, expression, purification, and screening of l-ASP recombinant enzymes
l-ASP expression vectors with a pelB leader sequence23 to facilitate secretion of recombinant l-ASP into the culture supernatant were transformed into the E coli BLR (DE3) strain (Novagen) (supplemental Figure 1). After using isopropyl β-D-1-thiogalactopyranoside (IPTG) to induce transformed cells to express and secrete l-ASP, we then screened for asparaginase and glutaminase activities using the aforementioned colorimetric assays. The enzymatically inactive T89V mutant24 and empty expression vector served as negative controls. Both asparaginase and glutaminase enzymatic activities were correlated with enzyme concentration (supplemental Figure 2). Hence, we successfully developed a rapid screening procedure for measuring the asparaginase and glutaminase activities of l-ASP without protein purification. To validate the method for use on unpurified supernatants, we performed parallel assays after purification of the l-ASP mutant proteins (supplemental Methods).
RNA interference (RNAi) and cell proliferation assays
All mammalian cell lines were maintained in RPMI 1640 (HyClone) with 5% fetal bovine serum (HyClone) and 2 mM l-glutamine (HyClone), as described previously.25 Small interfering RNA (siRNA) assays were performed as described previously25 with the following modifications: 96-well culture plates used final 5 nM siRNA, 0.10 µL of INTERFERin (PolyPlus Transfection), and 1500 cells per well in a 100-µL total volume. Cell proliferation was assessed using CellTiter-Blue or 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) assay (Promega) according to the manufacturer’s instructions, and l-ASP 50% effective concentration (EC50) values were determined using GraphPad Prism 6 (GraphPad Software) as described previously.25 All mammalian cell lines were tested for Mycoplasma using the MycoAlert assay (Lonza) at the commencement of this study and found to be negative. In addition, DNA fingerprints were obtained for all cell lines and were concordant with those that we reported previously.26
Detection of ASNS protein
For measurement of ASNS protein, total cell protein was extracted using bicine/CHAPs lysis buffer (Protein Simple). Twenty micrograms of total protein per lane was electrophoresed in sodium dodecyl sulfate (SDS)–polyacrylamide, transferred to Immun-Blot polyvinylidene difluoride membrane (Bio-Rad), and probed with antibodies against ASNS or β-actin (Sigma) as we reported previously.25 Relative ASNS and β-actin expression levels were quantified using ImageJ.
Kinetic competition analysis of l-ASP mutants
The kinetics of asparagine and glutamine catabolism by l-ASP were analyzed using a reaction containing 100 µM asparagine, 1600 µM glutamine, 23 mM Tris-HCl at pH 8.5 and either WT l-ASP (15 nM) or its Q59L mutant (200 nM). Aliquots (30 µL) were withdrawn from 500-µL reaction mixtures over a 60-minute time course. The reaction was immediately quenched by adding the aliquot to 120 µL of dry ice–cooled methanol containing 10 µM 13C,15N-labeled internal standards of asparagine, aspartate, glutamine, and glutamate. Those quenched aliquots were then analyzed by LC-MS/MS.
Results
MD of l-ASP
To guide mutagenesis experiments aimed at creating a glutaminase-deficient mutant, we performed 20-ns MD simulations of WT l-ASP bound with asparagine or glutamine. We compared preferential orientations and contacts of the 2 substrates within the lining of the l-ASP catalytic cleft and identified critical differences in how each substrate is coordinated. Analysis of enzyme-substrate contact times served as a basis for identifying the most promising mutagenesis target.
The simulation results were largely consistent with those published previously (reviewed in Labrou et al19 ), but one feature was surprising. Both substrates changed positions within ∼200 to 300 ps compared with the crystallographic orientation of aspartate. That re-orientation occurred in all 4 enzyme-binding pockets of the tetramer, clearly establishing a different preference for asparagine and glutamine compared with the product, aspartate. The re-orientation could be attributed to the fact that both substrates include uncharged amide side chains rather than the negatively charged carboxylate moiety of the product. Additional differences from crystal structures, which are formed by tightly packed enzymes under low hydration, are expected due to simulation conditions at physiological protein concentrations, ion concentrations, and temperature. The dynamics of equilibration in the course of 20-ns simulations is shown in supplemental Figure 3. As illustrated in supplemental Figure 3B, the probability of contact between glutamine side-chain amide oxygen (-CONH2, labeled “OE1”) and the enzyme diminished in the interval between 6 and 8 ns, whereas probabilities of other interactions fluctuated around stable mean values.
The enzyme residues and specific atoms forming the first shell around asparagine and glutamine are listed in Table 1 and supplemental Table 2. The criterion for inclusion in the first shell was proximity of <3 Å for a duration of >1% of the entire simulation time. Probabilities were averaged over all 4 of the enzyme’s binding sites. Table 1 and supplemental Table 2 suggest that both substrates are coordinated by essentially the same sets of atoms. Importantly, side-chain amide oxygens of asparagine and glutamine approached the catalytic hydroxyl (-OH) group of threonine T12 with probabilities of only 3% and 1%, respectively (supplemental Table 2). In contrast, their α-carboxylate groups (-COO−) contacted the catalytic -OH of T12 for much larger fractions of time (77% and 48%, respectively).
Polar groups of E colil-ASP that most frequently interact with the backbones of the substrates
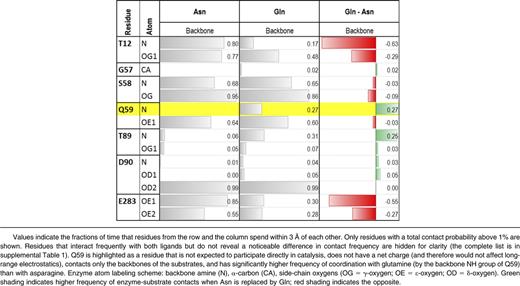
The differences between substrate contacts (Table 1 and supplemental Table 2; rightmost columns) indicated a decreased probability of contact between glutamine and almost every residue in the enzyme catalytic site. Notable exceptions were the backbone α-amino groups (-NH) of glutamine Q59 and threonine T89, and the side-chain amine (-NH3+) of lysine K162. The most notable difference between asparagine and glutamine substrates, in fact, was the mode of interaction with Q59 (Figure 1). Increased interaction of the glutamine substrate’s α-carboxyl with the Q59 backbone amide coincided with reduced interaction with the catalytic T12 residue. As a consequence, the α-carboxyl of glutamine re-oriented toward the backbone amide of T89, and the side-chain amide of glutamine lost contact with both the backbone amide and side-chain hydrogen bond donors of T89. As T89 typically coordinates the amide group of asparagine, the lost contact with glutamine appeared to be of significant importance.

Distinct coordinations of asparagine and glutamine in the catalytic site of E colil-ASP. Snapshots were taken at ∼20 ns of simulation. Q59 typically interacts with the backbone of both asparagine (A; light green) and glutamine (B; orange), but the patterns differ. Asparagine is usually coordinated through its backbone -NH group by the side-chain oxygen of Q59, whereas the backbone carboxyl of glutamine often interacts with the backbone -NH group of Q59, while the side chain of Q59 faces away from the substrate.
Distinct coordinations of asparagine and glutamine in the catalytic site of E colil-ASP. Snapshots were taken at ∼20 ns of simulation. Q59 typically interacts with the backbone of both asparagine (A; light green) and glutamine (B; orange), but the patterns differ. Asparagine is usually coordinated through its backbone -NH group by the side-chain oxygen of Q59, whereas the backbone carboxyl of glutamine often interacts with the backbone -NH group of Q59, while the side chain of Q59 faces away from the substrate.
Those results suggested that residue Q59 of l-ASP would be a more promising site for mutagenesis than K162 or T89. The latter residue plays an important catalytic role in the second stage of the reaction (hydrolysis of the aspartyl-enzyme bond) and, therefore, should not be mutated.24 Similarly, K162 is involved in electrostatic stabilization of several charges in the catalytic cleft24,27 and, thus, should not be mutated. Q59, on the other hand, coordinates the backbone groups but not the side chains of both substrates. Therefore, we reasoned that mutations in Q59 would be less likely to render the enzyme completely inactive.
Characterization of l-ASP Q59 mutants
To test experimentally the prediction that some mutations of residue Q59 would selectively decrease the glutaminase activity of l-ASP without rendering it enzymatically inactive, we performed site-directed mutagenesis to obtain Q59 variants of the enzyme. l-ASP expression vectors, coding for all 20 possible amino acids at position 59, were transformed into E coli BL-21. All mutants except Q59C and Q59S were expressed and secreted into the culture medium as efficiently as the WT protein (Figure 2A). Subsequent kinetic screening using colorimetric assays indicated that the mutants exhibited a spectrum of asparaginase activity ranging from 0% to 80% of WT, with a median of 12% (Figure 2B). The Q59 mutants also exhibited a spectrum of glutaminase activity ranging from 0% to 60% of WT, but the median was just 2% of WT glutaminase activity (Figure 2C), suggesting that Q59 is indeed more important for glutaminase activity than for asparaginase activity.

Enzymatic characterization of Q59 l-ASP mutants. (A) Coomassie blue–stained SDS-PAGE showing expression of l-ASP WT and Q59 mutants. The expression vector was transformed into E coli Bl-21 strain, and 20 µL of culture supernatant was analyzed by SDS-PAGE. The empty expression vector (Ctrl) and T89V (inactive mutant) served as negative controls for assays of enzyme activity in panels B and C. (B) Asparaginase activity of Q59 mutants by colorimetric assay. (C) Glutaminase activity of Q59 mutants by colorimetric assay. (D) Asparaginase-specific activity of purified Q59 mutants by the colorimetric assay. (E) Glutaminase-specific activity of purified Q59 mutants by the colorimetric assay. (F) Ratio of glutaminase- and asparaginase-specific activities for purified l-ASP mutants. SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
Enzymatic characterization of Q59 l-ASP mutants. (A) Coomassie blue–stained SDS-PAGE showing expression of l-ASP WT and Q59 mutants. The expression vector was transformed into E coli Bl-21 strain, and 20 µL of culture supernatant was analyzed by SDS-PAGE. The empty expression vector (Ctrl) and T89V (inactive mutant) served as negative controls for assays of enzyme activity in panels B and C. (B) Asparaginase activity of Q59 mutants by colorimetric assay. (C) Glutaminase activity of Q59 mutants by colorimetric assay. (D) Asparaginase-specific activity of purified Q59 mutants by the colorimetric assay. (E) Glutaminase-specific activity of purified Q59 mutants by the colorimetric assay. (F) Ratio of glutaminase- and asparaginase-specific activities for purified l-ASP mutants. SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electrophoresis.
To exclude the possibility that endogenous E colil-Asp was contributing to the asparaginase and glutaminase activities measured on nonpurified supernatants, we purified mutants Q59L, Q59F, Q59D, Q59E, Q59H, and Q59N and found that they yielded results (Figure 2D-E) consistent with the initial screen. That observation indicated that the screening of nonpurified supernatants was quantitatively reliable (supplemental Figure 2). Figure 2F illustrates glutaminase:asparaginase ratios of the purified mutants. At 1 extreme, Q59L and Q59F exhibited the smallest glutaminase:asparaginase ratios, with almost undetectable glutaminase activity yet 80% and 25% of WT asparaginase activity, respectively. Next, we used a sensitive LC-MS/MS assay to confirm that Q59L exhibits negligible glutaminase activity as indicated by measurement of glutamic acid after incubation with glutamine for 1 hour (supplemental Figure 4). For comparison, Q59L exhibited even lower glutaminase activity than that of W succinogenesl-Asp (supplemental Figure 4C), which was previously reported, using less sensitive methods, to exhibit very low glutaminase activity.15,28,29 At the other extreme, Q59H exhibited the largest ratio of glutaminase:asparaginase activity.
Kinetic characterization of Q59L l-ASP
We next compared Elspar, the clinical variant of l-ASP, with WT l-ASP and Q59 l-ASP with respect to their kinetics of asparagine and glutamine deamidation. Using optimized steady-state reaction conditions, we found that the initial rate of product formation (v0) measured by colorimetric asparaginase assay was equivalent for all 3 enzymes when used at equivalent asparaginase concentrations (supplemental Figure 5A-B). The corresponding glutaminase activity of WT l-ASP was slightly less than that of Elspar, and the glutaminase activity of Q59L was not detected.
We were concerned, however, that the colorimetric assays might be misleading for kinetic analysis because they are based on derivatives of the amino acid substrates and products, rather than the amino acids themselves. Hence, we turned to our LC-MS/MS assay for more reliable and sensitive analysis. First, to compare the glutaminase activities of the l-ASP variants, we determined that 10, 20, and 60 nM concentrations of WT, Q59L, and Q59H l-ASP, respectively, exhibited nearly identical asparaginase initial reaction rates (∼4.8 × 10−2 nmol/s) (supplemental Figure 5C). Using the same ratio (40, 80, and 240 nM) did not yield equivalent glutaminase activities; initial reaction rates were 1.7 × 10−3, <9.8 × 10−5 (near the assay detection limit), and 9.0 × 10−3 nmol/s, respectively (supplemental Figure 5D), indicating that Q59L exhibits undetectable glutaminase activity.
We next analyzed substrate competition kinetics using physiologically relevant concentrations of asparagine and glutamine. Figure 3 shows the resulting time course of asparagine depletion and glutamate formation in single-substrate reactions (black symbols) or in a mixture of the 2 substrates (white symbols). WT l-ASP completely degraded pure 100 µM asparagine in a linear fashion within ∼500 s (Figure 3A black circles), whereas the presence of 1600 µM glutamine delayed asparagine degradation to ∼600 s (Figure 3A, white circles). In the single reaction with glutamine, glutamate was formed immediately and linearly over 1200 s (Figure 3B, black triangles), but it was not detected in the mixture until ∼600 s (Figure 3B, white circles). As asparagine was almost fully depleted at 600 s in the reaction with both substrates (Figure 3A, open circles) and glutamate did not begin to appear until that point, the data suggest a strong kinetic preference of WT l-ASP for asparagine. Additional competition experiments in which both substrates were used at 1mM also yielded a time lag in the appearance of reaction products aspartic acid and glutamic acid (supplemental Figure 6). Q59L, to the contrary, did not exhibit measurable glutaminase activity (Figure 3B). Moreover, 1600 µM glutamine did not inhibit the asparaginase activity of Q59L, indicating that glutamine is not strongly bound (or deamidated) by Q59L.

Asparagine and glutamine deamidation kinetics. WT l-ASP or Q59L l-ASP was added to a reaction solution containing 100 µM asparagine, 1600 µM glutamine, or both. Concentrations of asparagine (A) and glutamic acid (B) were measured over a 1500-s time series by LC-MS/MS. Solid symbols represent concentrations in the single substrate reaction, and open symbols represent concentrations in the mixture.
Asparagine and glutamine deamidation kinetics. WT l-ASP or Q59L l-ASP was added to a reaction solution containing 100 µM asparagine, 1600 µM glutamine, or both. Concentrations of asparagine (A) and glutamic acid (B) were measured over a 1500-s time series by LC-MS/MS. Solid symbols represent concentrations in the single substrate reaction, and open symbols represent concentrations in the mixture.
Anticancer activity of l-ASP Q59 mutants
To investigate the contribution of glutaminase activity to the anticancer activity of l-ASP, we used purified WT, Q59L, or Q59F l-ASP to treat 6 leukemia lines (CCRF-CEM, SR, MOLT-4, K562, NALM-6, and REH) (Figure 4A-F) and 2 ovarian cancer lines (OVCAR-8 and SK-OV-3) (Figure 4G-H). Dosages were scaled to match asparaginase specific activity (IU/mg enzyme). The purified WT enzyme yielded anticancer activity comparable to that of Elspar (Figure 4A-B). Glutaminase-deficient Q59L and Q59F, to the contrary, did not exhibit measurable anticancer activity against any of the 8 lines, even at the highest dose, 32 U/mL, indicating that glutaminase activity is essential to the anticancer activity of l-ASP in those cell lines. In further support of that conclusion, W succinogenesl-ASP, which exhibits weak glutaminase activity (supplemental Figure 4C), retained only weak anticancer activity (supplemental Figure 7). In contrast to the glutaminase-deficient Q59L and Q59F mutants, Q59 mutants that retained glutaminase activity (Q59D, Q59E, Q59H, Q59N) exhibited anticancer activity comparable to that of WT l-ASP (Figure 4I-J). Overall, the results illustrated in Figure 4 indicate that the glutaminase activity of l-ASP contributes to anticancer activity in the cell lines listed in this section. However, in the next section, we show that glutaminase activity is not requisite for that activity in all cancer cell types.

Anticancer activity of WT, Q59L, and Q59F l-ASP. (A-H) Two ovarian cancer cell lines (OVCAR-8 and SK-OV-3) and 6 leukemia cell lines (MOLT-4, K562, NALM-6, REH, SR, and CCRF-CEM) were seeded in 96-well plates, incubated for 48 hours, treated with a range of (WT, Q59L, or Q59F) l-ASP concentrations for 48 hours, and finally assayed with CellTiter-Blue using fluorescence excitation at 544 nm and emission at 590 nm. (I-J) MOLT-4 and OVCAR-8 cells were seeded in 96-well plates and incubated for 48 hours, then treated with indicated concentrations of E colil-ASP WT or Q59 mutant for 48 hours. Inhibition of cell viability was measured as in panels A-H. Sham treatment was used as a control. (K) Western blot analysis of ASNS levels in the indicated cells treated with an EC50 dose of l-ASP. (L) Western blot analysis of ASNS levels in OVCAR-8 cells treated with l-ASP mutants. Numbers below the blot represent the relative level of ASNS, which was normalized to the level of the loading control β-actin (set to “1” for the control).
Anticancer activity of WT, Q59L, and Q59F l-ASP. (A-H) Two ovarian cancer cell lines (OVCAR-8 and SK-OV-3) and 6 leukemia cell lines (MOLT-4, K562, NALM-6, REH, SR, and CCRF-CEM) were seeded in 96-well plates, incubated for 48 hours, treated with a range of (WT, Q59L, or Q59F) l-ASP concentrations for 48 hours, and finally assayed with CellTiter-Blue using fluorescence excitation at 544 nm and emission at 590 nm. (I-J) MOLT-4 and OVCAR-8 cells were seeded in 96-well plates and incubated for 48 hours, then treated with indicated concentrations of E colil-ASP WT or Q59 mutant for 48 hours. Inhibition of cell viability was measured as in panels A-H. Sham treatment was used as a control. (K) Western blot analysis of ASNS levels in the indicated cells treated with an EC50 dose of l-ASP. (L) Western blot analysis of ASNS levels in OVCAR-8 cells treated with l-ASP mutants. Numbers below the blot represent the relative level of ASNS, which was normalized to the level of the loading control β-actin (set to “1” for the control).
ASNS mediates resistance to l-ASP Q59 mutants
We and others previously reported a negative correlation between the anticancer activity of l-ASP and the expression of ASNS.5,25,30 To determine whether ASNS also mediates resistance to glutaminase-deficient Q59 mutants, we first performed ASNS western blot analysis before and after treatment of cell lines that we had found to be insensitive to the glutaminase-deficient Q59L and Q59F l-ASP mutants. As expected, ASNS was extensively upregulated in all of the cell lines tested (Figure 4K). We next performed western blot analysis of ASNS following treatment of OVCAR-8 cells with selected Q59 mutants. ASNS was also upregulated by all 6 mutants tested, albeit to a different extent for each mutant (Figure 4L). Interestingly, high-glutaminase Q59H induced the lowest extent of ASNS upregulation, suggesting that the extent of ASNS upregulation may be suppressed by the glutaminase activity of l-ASP.
We next used a functional genomics approach, RNAi, to test the hypothesis that ASNS upregulation mediates resistance to glutaminase-deficient l-ASP. ASNS siRNA treatment resulted in highly effective knockdown of ASNS protein in OVCAR-8 cells (Figure 5A). In support of the hypothesis, ASNS siRNA potently sensitized OVCAR-8 cells to glutaminase-deficient Q59L (Figure 5B). Moreover, Q59L and Q59F exerted potent anticancer activity against the leukemia cell lines Sup-B15 and RS4;11 (Figure 5C-D), which did not express detectable levels of ASNS protein before or after l-ASP treatment (Figure 5E). Accordingly, we refer to those 2 cell lines as “ASNS-negative.” Notably, the anticancer activity of Q59L against the Sup-B15 line (EC50 = 1.4 × 10−4 U/mL) and RS4;11 (EC50 = 1.3 × 10−3 U/mL) was greater than or equal to the anticancer activity of Q59L against the ASNS siRNA-treated OVCAR-8 line (EC50 = 1.1 × 10−3 U/mL)—a degree of anticancer activity that reflects the greatest in vitro l-ASP potency we have observed to date while performing measurements on >70 cell types.5,6,25 In summary, these results demonstrate that ASNS-negative cancer cell lines are hypersensitive to asparaginase activity alone (ie, asparagine depletion without glutamine depletion).

Selective growth inhibition of ASNS-negative cancer cells by WT, Q59L, and Q59F l-ASP. (A) Western blot analysis of ASNS levels in OVCAR-8 cells after 48 hours transfection with ASNS siRNA (siASNS) or negative control siRNA (siNeg). β-actin was used as a loading control. (B) WT and Q59L l-ASP concentration-activity curves. The OVCAR-8 cell line was transfected with negative control siRNA (siNeg) or ASNS siRNA (siASNS) for 48 hours, then treated with a range of l-ASP concentrations for 48 hours, and finally assayed with CellTiter-Blue. WT, Q59L, and Q59F l-ASP concentration-activity curves were determined in the (C) Sup-B15 and (D) RS4;11 leukemia cell lines by CellTiter-Blue assay. (E) Western blot analysis of ASNS levels in Sup-B15 and RS4;11 cells treated with an EC50 dose of l-ASP. No treatment was used as a primary control, and MOLT-4 cells were included as a secondary control.
Selective growth inhibition of ASNS-negative cancer cells by WT, Q59L, and Q59F l-ASP. (A) Western blot analysis of ASNS levels in OVCAR-8 cells after 48 hours transfection with ASNS siRNA (siASNS) or negative control siRNA (siNeg). β-actin was used as a loading control. (B) WT and Q59L l-ASP concentration-activity curves. The OVCAR-8 cell line was transfected with negative control siRNA (siNeg) or ASNS siRNA (siASNS) for 48 hours, then treated with a range of l-ASP concentrations for 48 hours, and finally assayed with CellTiter-Blue. WT, Q59L, and Q59F l-ASP concentration-activity curves were determined in the (C) Sup-B15 and (D) RS4;11 leukemia cell lines by CellTiter-Blue assay. (E) Western blot analysis of ASNS levels in Sup-B15 and RS4;11 cells treated with an EC50 dose of l-ASP. No treatment was used as a primary control, and MOLT-4 cells were included as a secondary control.
Discussion
The enzyme-drug l-ASP has been used successfully for over 40 years to treat ALL. However, toxicity is a problem. Patients often cannot tolerate the 25-week course of l-ASP therapy that is frequently necessary to induce remission.10 The toxicity has been attributed to l-ASP’s glutaminase activity,8,9 but, to complicate matters, the anticancer activity has also been attributed to glutaminase activity.11-18 Hence, there are opposing views on how to improve the therapeutic index of l-ASP. Should glutaminase activity be increased or decreased? Our results suggest an unexpected answer—perhaps either approach could make sense, depending on ASNS expression in the target cancer.
Our first goal was to generate a glutaminase-deficient derivative of the clinically used E colil-ASP. The molecular structure of the E coli asparaginase active site in complex with aspartic acid was first revealed in the 3ECA x-ray crystal structure31 and later in a higher resolution structure, 1NNS.32 Those structures revealed contacts between S58, Q59, D90, E283, and backbone groups of aspartic acid. N246 and N248 did not contact either substrate directly, despite the possibility that N248 might stabilize E283 in proximity of the ligand. We observed similar contacts in our MD simulations, which identified Q59 as a catalytically nonessential residue with the greatest difference in contact pattern between the asparagine and glutamine substrates (Table 1 and Figure 1). Prior simulation studies (reviewed in Labrou et al19 ) have not focused on Q59, although experimental mutagenesis at residue Q59 yielded 3 mutants (Q59A, Q59E, and Q59G) that exhibited dramatically reduced enzyme activity.33 The relative neglect of Q59 in previous studies probably resulted from differences in the approaches used. The most relevant studies18,33 used crystallographic structures to identify targets, followed by MD simulations to explain the findings. Those investigations focused on intersubunit contacts and catalytic loop dynamics to find changes that might increase catalytic rate and enzyme stability. A recent simulation34 also studied the active site based on the 3ECA structure, which has only 1 water molecule in the active site, resulting in absent networks of hydrogen bonds that stabilize the active site. That paucity of water molecules could be significant. In contrast, we first performed MD simulations to hydrate and equilibrate the enzyme fully and, consequently, to explore its biologically relevant structures more fully. That approach changed the contact probabilities from those observed under enzyme-packed, low-hydration, crystallographic conditions, thereby changing the preferred mutagenesis targets.
In seeking a mutagenesis target that would suppress glutaminase activity but not asparaginase activity, we chose a residue with the following 4 characteristics: does not participate directly in catalysis, does not have a net charge, contacts only the backbone of the substrate, and preferentially contacts glutamine over asparagine. We eliminated N248 for lack of direct contact with the substrate and E283 for having charge. S58 and D90 satisfied 3 of the 4 criteria but contacted the 2 substrates with about equal frequency. Q59 satisfied all criteria.
Having identified Q59 as our leading target, we performed saturation mutagenesis of Q59 and developed a rapid colorimetric screening procedure to measure the asparaginase and glutaminase activities of the resulting mutants. Importantly, the experimental screening results corroborated the in silico predictions; mutation of Q59 generally decreased glutaminase activity to a greater extent than asparaginase activity (Figure 2). We further characterized some of the Q59 mutants using a sensitive LC-MS/MS assay. As a demonstration of the method’s sensitivity, we measured W succinogenesl-ASP glutaminase activity of 1.5 × 10−5 nmol/s (a level previously undetected by other methods) in the presence of 160 nM enzyme. Even with that sensitivity, however, Q59L showed no detectable glutaminase activity (supplemental Figure 4C). In addition, its affinity for glutamine was sufficiently low that even high concentrations of glutamine (up to 16 mM) did not inhibit its asparaginase activity (Figure 3A). That observation provides an attractive rationale for further consideration of Q59L as a drug candidate.
Because we and others have demonstrated that ASNS expression is correlated with resistance to l-ASP,3,5,6,25,30,35-40 studies of l-ASP must take ASNS expression into account. We first tested the anticancer activity of WT, Q59L, and Q59F l-ASP against 6 leukemia lines and 2 ovarian cancer lines that express ASNS. The glutaminase-deficient Q59L and Q59F l-ASP variants showed anticancer activity against ASNS-negative cell types (Sup-B15, RS4;11, and ASNS siRNA-treated OVCAR-8) (Figure 5) but not ASNS-positive cell types (Figure 4), supporting the central hypothesis of this study that the glutaminase activity of l-ASP is not always required for anticancer activity. Notably, “ASNS-positive” cell types included lines such as MOLT-4, for which baseline ASNS expression was almost undetectable yet was induced following l-ASP treatment, and lines such as K562, for which baseline ASNS expression was high (Figure 5K).
The model illustrated in Figure 6 summarizes our results. Cancer cells can be stratified into ASNS-negative and ASNS-positive, and the latter group can be further stratified into low and high ASNS. Only ASNS-positive cancer cells are able to use Gln imported from the extracellular environment to synthesize Asn, enabling them to proliferate regardless of the availability of extracellular Asn. Because Q59L l-ASP effectively depletes only Asn but not Gln, ASNS-positive cells are resistant to Q59L treatment (left panels, Figure 6A-B). In contrast, extracellular Asn is essential for proliferation of ASNS-negative cell types because of the inability to synthesize Asn endogenously (left panel, Figure 6C). WT l-ASP, however, depletes the extracellular supply of both Asn and Gln due to its added glutaminase activity. High-ASNS cancer cells may continue to proliferate following such treatment if intracellular synthesis of Asn and Gln are sufficient (right panel, Figure 6A). Low-ASNS cell types have reduced capacity to withstand such treatment, though; the reduced ability to produce Asn results in decreased cancer cell proliferation (right panel, Figure 6B). ASNS-negative cell types cannot withstand WT l-ASP treatment, and, importantly, l-ASP glutaminase activity appears to be unnecessary for inhibiting proliferation of ASNS-negative cells (right panel, Figure 6C). Additional studies will be required to more clearly define the thresholds (represented by horizontal dashed lines) that separate high-ASNS, low-ASNS, and ASNS-negative cell types, but it is nevertheless clear from our results that ASNS-negative cancer cells are hypersensitive to asparaginase treatment without glutaminase activity.

Proposed model for the mechanism of WT l-ASP’s and Q59L lASP’s anticancer activity. The mechanism of anticancer activity depends on l-ASP glutaminase activity and ASNS expression, which is reflected by the color gradient of the background. For simplicity, glutamine synthesis pathways are not shown. (A) (Left panel) (1) Q59L l-ASP effectively depletes Asn but not Gln, which (2) is imported by the cancer cell for (3) synthesis of Asn by ASNS, thereby promoting cancer cell proliferation (4). Numbering is omitted from subsequent panels, but analogous interpretation illustrates that the added glutaminase activity of WT l-ASP decreases the extracellular supply of Gln, thereby limiting cancer cell proliferation (right panel). (B) Low-ASNS cancer cells are insensitive to Q59L l-ASP (left panel), but not to WT l-ASP (right panel). (C) ASNS-negative cancer cells are sensitive to both Q59L (left panel) and WT (right panel). Details of the model are provided in the text.
Proposed model for the mechanism of WT l-ASP’s and Q59L lASP’s anticancer activity. The mechanism of anticancer activity depends on l-ASP glutaminase activity and ASNS expression, which is reflected by the color gradient of the background. For simplicity, glutamine synthesis pathways are not shown. (A) (Left panel) (1) Q59L l-ASP effectively depletes Asn but not Gln, which (2) is imported by the cancer cell for (3) synthesis of Asn by ASNS, thereby promoting cancer cell proliferation (4). Numbering is omitted from subsequent panels, but analogous interpretation illustrates that the added glutaminase activity of WT l-ASP decreases the extracellular supply of Gln, thereby limiting cancer cell proliferation (right panel). (B) Low-ASNS cancer cells are insensitive to Q59L l-ASP (left panel), but not to WT l-ASP (right panel). (C) ASNS-negative cancer cells are sensitive to both Q59L (left panel) and WT (right panel). Details of the model are provided in the text.
The glutaminase activity of l-ASP has been implicated in many ALL treatment-associated side effects including immune suppression, pancreatitis, liver damage, and neurotoxicity.9,41-45 A potential strength of glutaminase-deficient l-ASP variants is, therefore, the possibility of improved therapeutic index if the modified l-ASP remains active against the cancer cells. For example, glutaminase-free l-ASP variants such as Q59L might not induce pancreatitis. Reports that glutamine supplementation is highly effective in treating pancreatitis46 provide further support for that speculation. Nevertheless, preclinical studies using animal models will be critical for determining whether Q59L exhibits improved therapeutic index.
We noted a minor inconsistency between our results and the literature. A different glutaminase-deficient variant of l-ASP (N24A, Y250L double mutant) was reported to exhibit anticancer activity against the REH cell line,18 which we found to be highly resistant to Q59L and Q59F l-ASP. Because those authors did not measure ASNS expression or perform DNA fingerprinting analysis of their cell lines, we cannot be sure that our REH lines were indeed the same. Additionally, they did not report full dose-response curves, so we cannot confirm that the methods for measuring anticancer activity were similar. Another minor discrepancy with that same report is that our Sup-B15 line was much more sensitive to glutaminase-deficient l-ASP variants. Nevertheless, our results were highly consistent with those recently reported by another group that showed Sup-B15 to be sensitive to asparagine depletion alone without glutamine depletion.12
In conclusion, we report (1) that the glutaminase activity of l-ASP is necessary for anticancer activity against cancer cells that express significant ASNS but (2) that ASNS-negative cancer cells are highly sensitive to asparaginase activity alone. Because the glutaminase activity of l-ASP is believed to be responsible for its toxicity, these findings provide a rationale for testing the hypothesis that a glutaminase-deficient l-ASP variant (eg, Q59L) will exhibit greater therapeutic index than that of WT l-ASP against ASNS-negative cancers. Overall, our results support both sides of the debate over increasing vs decreasing the glutaminase activity of l-ASP. That is, decreasing glutaminase activity may be sufficient to treat ASNS-negative cancers, whereas increasing glutaminase activity appears to be necessary for the treatment of ASNS-positive cancers.
There is an Inside Blood Commentary on this article in this issue.
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Acknowledgments
We thank David Hawke, Director of the Proteomics and Metabolomics Core Facility at Anderson Cancer Center, for assistance with mass spectrometry.
This work was supported in part by US National Cancer Institute at the National Institutes of Health grant numbers CA143883 and CA083639, the Cancer Prevention Research Institute of Texas grant number RP130397, the Chapman Foundation, and the Michael and Susan Dell Foundation (honoring Lorraine Dell). This work was also supported by Sandia’s Laboratory Directed Research and Development program. Sandia National Laboratories is a multiprogram laboratory operated by Sandia Corp, a wholly owned subsidiary of Lockheed Martin Corp, for the US Department of Energy National Nuclear Security Administration under contract DE-AC04-94AL85000.
Authorship
Contribution: W.K.C., P.L.L., and J.N.W. conceived and designed the research and analyzed data; W.K.C. performed most of the experiments, with technical assistance from P.P.; A.A. performed most of the simulations; A.A., D.M.R., S.S., and S.B.R. designed simulations and analyzed data; and W.K.C., P.L.L., A.A., D.M.R., S.S., S.B.R., and J.N.W. wrote the paper.
Conflict-of-interest disclosure: P.L.L. declares a conflict of interest based on consultancy and membership on the scientific advisory board of a company that markets an l-ASP product. P.L.L. and J.N.W. declare a conflict of interest based on patents and royalties related to l-ASP. All authors declare a conflict of interest related to a submitted patent application.
The current affiliation for D.M.R. is Department of Chemistry, University of South Florida, Tampa, FL 33620.
Correspondence: Philip L. Lorenzi, MD Anderson Cancer Center, 7435 Fannin St, Room 2SCR3.3027, Unit 0950, Houston, TX 77054; e-mail: PLLorenzi@mdanderson.org.
