Abstract
Hemophagocytic lymphohistiocytosis (HLH) is a syndrome describing patients with severe systemic hyperinflammation. Characteristic features include unremitting fever, cytopenias, hepatosplenomegaly, and elevation of typical HLH biomarkers. Patients can develop hepatitis, coagulopathy, liver failure, central nervous system involvement, multiorgan failure, and other manifestations. The syndrome has a high mortality rate. More and more, it is recognized that while HLH can be appropriately used as a broad summary diagnosis, many pediatric patients actually suffer from an expanding spectrum of genetic diseases that can be complicated by the syndrome of HLH. Classic genetic diseases in which HLH is a typical and common manifestation include pathogenic changes in familial HLH genes (PRF1, UNC13D, STXBP2, and STX11), several granule/pigment abnormality genes (RAB27A, LYST, and AP3B1), X-linked lymphoproliferative disease genes (SH2D1A and XIAP), and others such as NLRC4, CDC42, and the Epstein-Barr virus susceptibility diseases. There are many other genetic diseases in which HLH is an infrequent complication of the disorder as opposed to a prominent manifestation of the disease caused directly by the genetic defect, including other primary immune deficiencies and inborn errors of metabolism. HLH can also occur in patients with underlying rheumatologic or autoinflammatory disorders and is usually designated macrophage activation syndrome in those settings. Additionally, HLH can develop in patients during infections or malignancies without a known (or as-yet-identified) genetic predisposition. This article will attempt to summarize current concepts in the pediatric HLH field as well as offer a practical diagnostic and treatment overview.
Introduction: HLH and terminology
This review aims to describe the mechanisms, clinical presentation, biomarkers, and various treatments associated with a complex, life-threatening systemic hyperinflammatory syndrome called hemophagocytic lymphohistiocytosis (HLH). This syndrome is characterized by unremitting fever, cytopenias, hepatosplenomegaly, coagulopathy, and elevations in typical biomarkers, including ferritin and soluble interleukin-2 (IL-2) receptor (sIL-2R). Patients can also develop rash, hepatitis, disseminated intravascular coagulation, acute liver failure, central nervous system (CNS) involvement, multiorgan failure, and other problems, too often including death. The high mortality rate makes prompt recognition and treatment of this hyperinflammatory syndrome essential. Though imperfect, for the purposes of this review, we will use the term HLH to refer to this syndrome in general, irrespective of context or genetic susceptibility. A host of other umbrella terms have been employed for this purpose, including hyperinflammation, HLH syndrome, HLH-spectrum disorder, hyperferritinemic inflammation, and cytokine storm. Further, we will make use of a variety of other terms to connote specific contexts/etiologies of HLH (summarized in Table 1).
The first description of an inherited form of HLH, called familial hemophagocytic reticulosis, was in 1952.1 The first genetic discoveries of predisposition to HLH came in the late 1990s with the breakthrough findings of pathologic variants in LYST, SH2D1A, and PRF1.2-6 Since that time, the number and scope of additional genetic susceptibilities to HLH that have been discovered have revolutionized the field. It is imperative to make a distinction between genetic HLH disorders and the syndrome of HLH, as patients with many of the genetic diseases that cause HLH may be optimally treated by allogeneic hematopoietic cell transplantation (HCT). Several genetic diseases that feature HLH as the predominant manifestation are grouped as familial HLH and include pathologic changes in PRF1, UNC13D, STX11, and STXBP2 (familial HLH2-HLH5, respectively). HLH is also a common manifestation of several other genetic diseases, including certain pigmentary disorders, X-linked lymphoproliferative diseases, Epstein-Barr virus (EBV) susceptibility disorders, certain CDC42 mutations, and activating mutations in NLRC4. All of these disorders can be collectively termed genetic HLH diseases, and the term primary HLH can also be used as a category for some or all of these diseases.7
Many other primary immune deficiency (PID) disorders can be rarely complicated by HLH, usually in the setting of infections, and several inborn errors of metabolism can also be complicated by HLH, presumably due to abnormal macrophage activation. HLH can be referred to as secondary HLH in these settings.7 HLH is infrequent in these disorders and occurs as a complication of the diseases (eg, severe adenovirus infection in a patient with severe combined immune deficiency [SCID]) as opposed to being a primary manifestation of the disease itself. Secondary HLH is also used to describe HLH that is associated with an infection, malignancy, or autoimmune disease in the absence of any frank genetic predisposition to HLH.7 When HLH or a syndrome resembling HLH occurs in the setting of a rheumatologic or autoinflammatory disease, it is often referred to as macrophage activation syndrome (MAS).8 HLH in this setting can also be said to be caused by autoinflammation, implying innate immune activation is an important primary driver in this context.
Diagnosis of the syndrome of HLH
The Histiocyte Society established a set of clinical and laboratory criteria to help formalize the diagnosis of the syndrome of HLH for its HLH-94 and HLH-2004 clinical trials.9-11 A diagnosis of HLH was met in the HLH-2004 study if patients had 5 out of 8 clinical criteria, and many physicians still use these criteria when considering a diagnosis of the syndrome of HLH. The most recent criteria include fever, splenomegaly, cytopenias, hypertriglyceridemia and/or hypofibrinogenemia, hemophagocytosis, decreased natural killer (NK)-cell function, elevated ferritin, and elevated soluble IL-2 receptor levels (Table 2). CNS involvement was not included in the criteria; however, it occurs in 30% to 73% of HLH patients, and can include seizures, focal deficits, meningismus, and altered levels of consciousness; and portends worse outcomes.12
These criteria help guide diagnosis, but clinicians should not be overly strict given the time it takes to receive some results. Moreover, meeting HLH criteria does not indicate the absence of underlying infection or malignancy. The diagnostic criteria reflect inherited susceptibility to HLH (NK function), immune activation (ferritin and sIL-2R), and immunopathology (hemophagocytosis, splenomegaly, and disseminated intravascular coagulation) that reflect the extreme inflammation of HLH. Early disease presentations may not have progressed to have 5 out of 8 criteria yet, and some patients may never meet criteria, including those with atypical presentations such as isolated CNS disease or acute liver failure. In fact, as many as 17% of patients with proven genetic HLH diseases have been observed to have incomplete or atypical presentations.13 Isolated CNS HLH can be extremely challenging to diagnose and has been observed more commonly in older patients with hypomorphic mutations.14-19
It is particularly worth mentioning that the “pathognomonic” observation of hemophagocytosis is not required for a diagnosis and has poor sensitivity and specificity.20-22 The observation of decreased NK-cell function additionally has limitations. Very low or absent function can indicate a genetic HLH disease associated with compromised lymphocyte cytotoxicity. However, acute illness and various treatments can temporarily impair NK numbers and function, and a low result was found to have poor specificity (43%) by a large tertiary referral laboratory.23 Regardless of its diagnostic accuracy in screening for genetic cytotoxic defects (discussed later), the NK-cell function test does not help when considering if a patient has the syndrome of HLH and should primarily be used as a screening test for familial HLH and related pigmentary disorders that cause defective cytotoxicity.
Several laboratory tests are used to help in the diagnosis of the syndrome of HLH and are also useful biomarkers for disease activity monitoring (Figure 1). As previously mentioned, blood levels of triglycerides, fibrinogen, ferritin, and sIL-2R are all part of the criteria often used to diagnose a syndrome of HLH. Some degree of ferritin elevation is essentially required for the diagnosis of HLH. However, serum ferritin is also driven by iron overload states like sickle-cell disease and can be elevated in many inflammatory contexts. A level >500 µg/L is >90% sensitive, but specificity is only robust at levels >2000 to 10 000 µg/L, and in adults, levels >10 000 µg/L are still most commonly associated with malignancy.11,24,25 A practical analysis was performed by Lehmberg at al comparing 123 patients with HLH to 320 patients with other hyperferritinemic conditions. Their “trade-off” cut point of 2000 µg/L offered a sensitivity of 70% and specificity of 68% for HLH.26 The clinical sensitivity of sIL-2R can be estimated at 88% to 100%, depending on the population studied and sIL-2R cutoff used.27,28 It was elevated in 97% of patients enrolled in HLH-2004 using ≥2400 U/mL as the cutoff.11 Like ferritin, an elevated sIL-2R alone does not indicate HLH, though very high levels seem to be associated with profound T-cell activation. Additionally, normal ranges are age dependent. Importantly, it can be elevated in lymphoma, which is sometimes missed during the evaluation of HLH; a sIL-2R/ferritin ratio may be helpful to identify patients with lymphoma.29,30 Some centers use flow cytometric evaluation of T-cell HLA-DR expression (a marker of T-cell activation) in place of or in addition to measurements of sIL-2R.31 HLA-DR expression by >14.4% of CD8+ T cells and >4.8% of CD4+ T cells each had >80% sensitivity and 90% specificity for HLH in a German cohort.

Representative nonexhaustive flowchart of testing that can be considered during the evaluation of a pediatric patient with suspected HLH. No one algorithm is right for every patient. Clinicians should always consult with local multidisciplinary specialists regarding evaluations and consider consultation with physicians with expertise in HLH. Genetic testing should also be performed in many cases. CBC, complete blood count; CMV, cytomegalovirus; HSV, herpes simplex virus; PCR, polymerase chain reaction; PET-CT, positron emission tomography-computed tomography; TB, tuberculosis; TREC, T-cell receptor excision circle; VZV, varicella-zoster virus.
Representative nonexhaustive flowchart of testing that can be considered during the evaluation of a pediatric patient with suspected HLH. No one algorithm is right for every patient. Clinicians should always consult with local multidisciplinary specialists regarding evaluations and consider consultation with physicians with expertise in HLH. Genetic testing should also be performed in many cases. CBC, complete blood count; CMV, cytomegalovirus; HSV, herpes simplex virus; PCR, polymerase chain reaction; PET-CT, positron emission tomography-computed tomography; TB, tuberculosis; TREC, T-cell receptor excision circle; VZV, varicella-zoster virus.
For rheumatologists who encounter HLH/MAS in the setting of known or suspected systemic juvenile idiopathic arthritis, criteria proposed by the European League Against Rheumatism, American College of Rheumatology, and Pediatric Rheumatology International Trials Organization are often used (Table 3).32 The criteria are similar to features used by the Histiocyte Society for their clinical trials but are fewer and have different cutoffs based on detailed comparisons of patients with systemic juvenile idiopathic arthritis and MAS to patients with systemic juvenile idiopathic arthritis without MAS and patients with systemic infection. For example, fever was observed in >98% of patients with MAS but also in >94% of patients with either active systemic juvenile idiopathic arthritis or systemic infection.32 Criteria that highly differentiated MAS became part of the classification criteria. Per the criteria, any patient with known or suspected systemic juvenile idiopathic arthritis would be classified as having MAS with an elevated ferritin >684 ng/mL plus 2 of the following: platelet count ≤181 × 109/L, aspartate aminotransferase >48 U/L, triglycerides >156 mg/dL, and fibrinogen ≤360 mg/dL.32
Newer clinical testing options such as IL-18 levels, which reflect inflammasome activation, or CXCL9, which indicates interferon-γ (IFN-γ) pathway activity, are being more frequently used. There has not been much published to date regarding diagnostic accuracy, but more evidence will likely be forthcoming. CXCL9 has been used to indicate and follow IFN-γ activity in trials of an IFN-γ neutralizing antibody (emapalumab) in HLH and MAS (data leading to US Food and Drug Administration approval are available at https://www.accessdata.fda.gov/drugsatfda_docs/nda/2018/761107Orig1s000MultidisciplineR.pdf). The ratio of IL-18 to CXCL9 has been used to differentiate patients with rheumatologic diseases and MAS from patients with HLH.33 A ratio of total IL-18/CXCL9 of <2.3 was able to differentiate all patients with familial HLH from patients with MAS.33 Alone, a total IL-18 level >24 000 pg/mL distinguished MAS from familial HLH with 83% sensitivity and 94% specificity; levels were also higher in MAS than in patients with infection-associated HLH.33 Though IL-18 was starkly elevated in MAS, its natural inhibitor, IL-18 binding protein (IL-18BP), was more elevated in FHL and malignancy-associated HLH, and “free” IL-18 was associated with MAS susceptibility. IL-18BP may be another promising biomarker of IFN-γ activity. This supports a notion that autoinflammatory and rheumatologic diseases with MAS derive from a primary inflammasome/macrophage activation state with secondary T-cell activation, whereas patients with familial HLH disorders may have disease driven by pathologic T-cell activation. Balancing/complicating this hypothesis is the repeated observation that heterozygous FHL mutations, while common in databases of healthy volunteers, seem to be enriched in MAS populations.34-36 IL-18 levels can also be helpful when evaluating patients for XIAP deficiency or NLRC4 mutations, as patients with XIAP deficiency maintain elevated levels even during times of wellness due to dysregulated NLRP3 inflammasome activation,37-39 and patients with activating NLRC4 mutations usually have extremely high levels of IL-18 (near 100 000).40,41 Measurement of several other cytokines can be helpful in the diagnosis of the syndrome of HLH. In particular, IFN-γ and IL-10 have been observed to be elevated in patients with HLH as compared with controls and to patients with sepsis.42,43 One group observed that using a cut point of 100 pg/mL for IFN-γ levels yielded 94% sensitivity and 97% specificity for HLH, and the diagnostic accuracy of IL-10 levels was also good.43
Pathophysiology of HLH
Our understanding of HLH has been greatly advanced in the last 20 years thanks to the many genetic discoveries that have been made. Genes that are critical for either lymphocyte cytotoxicity or inflammasome activity are most often compromised in patients with primary HLH (Table 4 and Figure 2).
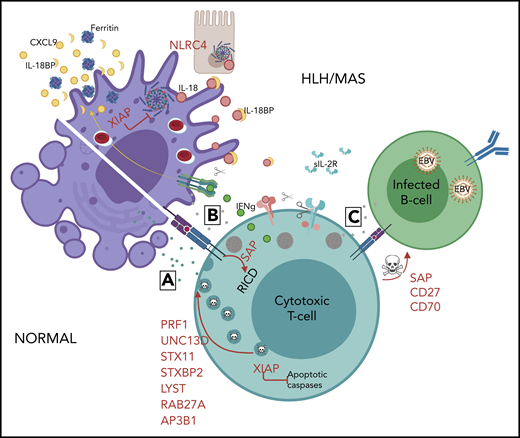
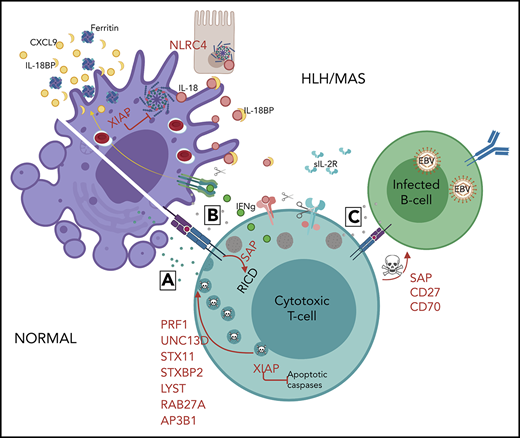
Mechanisms of genetic HLH predisposition. HLH is thought to develop due to abnormal reciprocal activation of mononuclear phagocytes (MNPs; monocytes, macrophages, and dendritic cells) and type 1 lymphocytes (NK cells and Th1, CD8, and NKT cells). T cells with normal cytotoxic granules release them to induce MNP apoptosis and terminate the synapse, whereas impaired or perforin-deficient granules (gray) cannot terminate MNP activation. Immune synapse prolongation and excess IL-18 (from MNP and/or epithelial sources) both amplify production of lymphocyte cytokines like IFN-γ, which in turn further activates MNPs and promotes hemophagocytosis and release of HLH biomarkers like ferritin, CXCL9, and IL-18BP. Activated lymphocytes upregulate the IL-2 receptor, which is cleaved by proteases released by activated MNPs. The absence of XIAP may permit pathogenic inflammasome activation and lymphocyte apoptosis, while SAP deficiency impairs restimulation-induced cell death (RICD) and, like CD27 and CD70 deficiency, prevents normal killing of EBV-infected B cells.
Mechanisms of genetic HLH predisposition. HLH is thought to develop due to abnormal reciprocal activation of mononuclear phagocytes (MNPs; monocytes, macrophages, and dendritic cells) and type 1 lymphocytes (NK cells and Th1, CD8, and NKT cells). T cells with normal cytotoxic granules release them to induce MNP apoptosis and terminate the synapse, whereas impaired or perforin-deficient granules (gray) cannot terminate MNP activation. Immune synapse prolongation and excess IL-18 (from MNP and/or epithelial sources) both amplify production of lymphocyte cytokines like IFN-γ, which in turn further activates MNPs and promotes hemophagocytosis and release of HLH biomarkers like ferritin, CXCL9, and IL-18BP. Activated lymphocytes upregulate the IL-2 receptor, which is cleaved by proteases released by activated MNPs. The absence of XIAP may permit pathogenic inflammasome activation and lymphocyte apoptosis, while SAP deficiency impairs restimulation-induced cell death (RICD) and, like CD27 and CD70 deficiency, prevents normal killing of EBV-infected B cells.
NK cells and cytotoxic T lymphocytes eliminate virus-infected or malignant cells using a variety of mechanisms, including several that are dependent on cytotoxic granule–mediated killing. Granule-mediated cytotoxicity is accomplished via the polarized delivery of cytotoxic granule contents to the immunologic synapse, extrusion of contents into the space shared with a target cell, and perforin-mediated entry into the target cell where cytotoxic granule contents induce target-cell apoptosis. This complex process has been studied and described in detail.44-47 In patients with familial HLH2 to HLH5 and the pigmentary disorders associated with HLH, granule-mediated cytotoxicity is impaired and is thought to lead to HLH by multiple mechanisms. Defects resulting in more severe impairment have been associated with earlier HLH onset and more severe disease.48 A prolonged synapse time occurs between cytotoxic lymphocytes that are deficient in perforin or granzymes and target cells and leads to overproduction of inflammatory cytokines.49 Antigen-presenting cells accumulate and continue to stimulate T cells, which further escalates T-cell activation and proliferation. A vicious cycle of continuing lymphohistiocytic proliferation and hypercytokinemia ensues, which ultimately leads to widespread tissue damage and the life-threatening hyperinflammatory syndrome of HLH. Some mechanisms of normal immune homeostasis are also hindered by defective granule-mediated cytotoxicity such as NK-cell–mediated elimination of CD4+ T cells, elimination of CTLs by granule-containing T-regulatory cells, and T-cell fratricidal processes, which further contribute to HLH.50
For patients who have defects in genes that compromise inflammasome regulation, including gain-of-function variants in NLRC4 and deficiency of XIAP, HLH is thought to be ignited by overactive macrophages and other cells that overproduce inflammasome-dependent cytokines. IL-18 may be specifically associated with MAS susceptibility, as many autoinflammatory diseases with profound IL-1 overproduction have not been associated with hyperinflammation.33 The resultant HLH can be indistinguishable from patients with granule-mediated cytotoxicity defects.
Genetic causes of HLH
Familial HLH and pigmentary disorders
There are many genetic causes of predisposition to HLH (Table 4 and Figure 2). The first genetic cause of familial HLH to be discovered was PRF1.2 PRF1 encodes perforin, which is contained within the cytotoxic granules of cytotoxic lymphocytes. Once released from the granules within the immunologic synapse, perforin oligomerizes on target cells to form pores that allow entry of other cytotoxic granule contents into the target cell, where they induce apoptosis. In patients with perforin deficiency, granule contents cannot enter target cells, which leads to a pathophysiologic setup for HLH as discussed above. Patients with perforin deficiency can be classified as having familial HLH type 2. The cause of familial HLH type 1 remains to be elucidated. Familial HLH types 3 to 5 are due to mutations in UNC13D, STX11, and STXBP2, respectively.51-54 The protein products of these genes are critical for normal cytotoxic granule exocytosis. In patients with defects in these genes, granule contents do not get released into the immunological synapse, and target cells are not able to be killed.
The pigmentary disorders that are associated with HLH have more widespread compromise of granule trafficking that can also (variably) affect melanocyte, platelet, neutrophil, and/or other granule release. Mutations in RAB27A, LYST, and AP3B1 cause Griscelli syndrome type 2, Chediak-Higashi syndrome, and Hermansky-Pudlak syndrome type 2, respectively.3,55,56 Notably, patients with Hermansky-Pudlak syndrome type 2 have a lower incidence of HLH than other disorders. Depending on the syndrome, patients can also have bleeding tendencies due to platelet granule dysfunction, neutropenia, and progressive neurodevelopmental abnormalities.
XLP1 and XLP2
The X-linked lymphoproliferative (XLP) diseases caused by pathologic variants in SH2D1A (XLP1) and XIAP (XLP2) cause HLH in addition to other clinical manifestations.4-6,57 SH2D1A encodes SLAM-associated protein (SAP), which is a small adaptor protein that is critical for regulating the signaling of the SLAM family of receptors. Lack of SAP leads to defective 2B4-mediated cytotoxicity (which is important for cytotoxicity against EBV-infected B cells), absent development of invariant NKT cells, and resistance to T-cell restimulation–induced cell death, which is important for normal T-cell response downregulation.58-61 These problems all contribute to the pathologic predisposition to HLH, which is almost exclusively associated with EBV in patients with XLP1. XLP1 patients also commonly develop lymphoma and humoral deficiency. They more rarely develop vasculitis or aplastic anemia.
XIAP deficiency causes a complex disease. Most relevant to HLH, deficiency of XIAP leads to dysregulated NLRP3 inflammasome activity with overproduction of inflammatory IL-1β and IL-18.39 Chronically elevated levels of IL-18 can be found in these patients and likely significantly contribute to the HLH susceptibility.37 XIAP also directly inhibits caspase-3, caspase-7, and caspase-9 and regulates cell death.62 XIAP-deficient cells, including T cells from XIAP-deficient patients, have an increased susceptibility to cell death, which may also play into HLH pathogenesis due to inefficient effector cell function.57 Less commonly than with SAP deficiency/XLP1, XIAP-HLH is triggered by EBV infection in 30% to 80%.63,64 Patients with XIAP deficiency do not develop lymphoma, but they commonly develop inflammatory bowel disease, hypogammaglobulinemia, recurrent infections, and other less common complications such as uveitis, periodic fever, granulomatous, lymphocytic interstitial lung disease, and fistulating skin disease, among other complications.57,63-67 Notably, disease manifestations have been reported in females with skewed X chromosome lyonization.68-70
NLRC4
Gain-of-function mutations in NLRC4 were discovered to cause HLH and enterocolitis by 2 groups in 2014, which was subsequently confirmed by many other groups.40,41 Mutations can be dominantly inherited or occur as de novo germline or even high-frequency somatic mutations.71 The activating mutations in NLRC4 lead to constitutive activation of the NLRC4 inflammasome, and patients with activating NLRC4 mutations have very high blood levels of IL-18, which is thought to play a significant role in disease pathogenesis. Patients may respond to recombinant IL-18BP, which sequesters IL-18 and prevents its activity.72 A clinical trial of IL-18BP (tadekinig alfa) for patients with mutations in NLRC4 or XIAP is ongoing (NCT03113760).
CDC42
Heterozygous mutations in CDC42 that affect the protein at C-terminal amino acids 186, 188, or 192 were very recently discovered to cause autoinflammation in 8 patients, including fatal HLH in 3 patients.73,74 Neonatal cytopenias, hepatosplenomegaly, transaminitis, recurrent febrile episodes, urticaria-like rashes, failure to thrive, MAS/HLH, and facial dysmorophisms were common characteristics in patients, along with elevated inflammatory markers. These mutations are distinct from other CDC42 mutations that have been described to cause Takenouchi-Kosaki syndrome, a diverse syndrome characterized by variable developmental delays, facial dysmorphism, cardiac or brain abnormalities, and hematologic and immunologic abnormalities.75,76 The described mutations in CDC42 are postulated to interfere with the binding and localization of CDC42 and interfere with normal actin assembly, thus affecting normal signaling, cytoskeletal rearrangement, polarization, proliferation, migration, and cytotoxicity processes. Although CDC42 is ubiquitously expressed, patients have very high levels of IL-18 and increased production of IL-1β ex vivo, suggesting dysregulated inflammasome function and an autoinflammatory nature of disease.73,74 CXCL9 levels were also elevated in some patients. One group summarized this disease as an IL-1 inhibition–responsive autoinflammatory syndrome.74 Patients have been treated with IL-1β inhibitors and corticosteroids, and 1 patient received treatment with emapalumab for a severe HLH episode, which resolved. One patient successfully underwent allogeneic HCT.73
EBV susceptibility diseases
Beyond XLP1, there are several PIDs that make patients highly susceptible to problems with EBV, including severe EBV, EBV-HLH, chronic active EBV, and lymphomas. These disorders should be considered in the differential diagnosis of patients with EBV-HLH if the more classic disorders are not found, and several can be considered as primary/genetic HLH diseases. Genes to evaluate include MAGT1, ITK, CD27, CD70, CTPS1, and RASGRP1 (Table 4). The diseases caused by mutations in these genes are complex and were recently reviewed by Latour and Winter.77
PIDs and inborn errors of metabolism
Several PIDs have been observed to be rarely complicated by HLH (Table 5), usually in the setting of infection. A large survey and literature search performed by the Histiocyte Society, the European Society of Blood and Bone Marrow Transplantation’s Inborn Errors Working Party, and the German Society for Pediatric Oncology and Hematology identified >60 patients with PIDs other than familial HLH, pigmentary disorders, or XLP who developed HLH.78 Most patients had chronic granulomatous disease or severe SCID, and there were also patients with various CIDs such as Wiskott-Aldrich syndrome, DiGeorge syndrome, ataxia telangiectasia, and other diagnoses, including X-linked agammaglobulinemia and autoimmune lymphoproliferative disease syndrome.78 Notably, many SCID and CGD patients presented with HLH prior to the diagnosis of SCID or CGD, and clinicians should bear this in mind when evaluating patients with HLH. Almost all patients with SCID and CID presented with virus-associated HLH, including EBV, cytomegalovirus, and adenovirus, among others.78 Patients with CGD most commonly presented with HLH associated with Burkholderia cepacia, Leishmania species, and fungi.78 Notably, a large single center reported the identification of PIDs in 14 out of 47 pediatric patients (30%) with HLH who lacked a genetic HLH disease.79 Diseases included those mentioned above plus DOCK8, STAT1, STAT2, STAT3, and PIK3CD (Table 5). The high percentage of PID patients found in this cohort suggests that whole-exome sequencing or large PID panel testing should be performed for patients with HLH for whom an HLH-causing disease is not found. Interestingly, HLH can also occur in the setting of IFN-γ receptor deficiency and was the presenting clinical phenotype in 2 patients with disseminated mycobacterial disease due to underlying IFN-γR1 or IFN-γR2 deficiency.80
Several inborn errors of metabolism can also present with HLH, particularly lysinuric protein intolerance due to mutations in SLC7A7 (Table 5).81-93 The accumulation of nondegraded substrates in macrophages may lead to inflammasome activation that triggers uncontrolled macrophage activation and the subsequent development of HLH.
Disease triggers and secondary HLH
Infection and malignancy, acting alone or in concert with the above-mentioned genetic susceptibility factors, are common HLH triggers.94,95 HLH that occurs in a patient who has a strong immunologic trigger such as infection or malignancy, in the absence of a genetic disease which features HLH, are often said to have secondary HLH. HLH can occur with virtually any infection or malignancy. More common infections include DNA viruses (EBV, cytomegalovirus, and adenovirus) and intracellular pathogens (eg, Leishmania), but the list of infections that have been reported to occur with HLH is extensive and influenced by geographic region (leishmaniasis and tick-borne illnesses), season (influenza viruses, tick-borne illnesses), and socioeconomic status (tuberculosis). Immune reconstitution inflammatory syndrome occurring when newly treated HIV patients are coinfected with (usually) tuberculosis bears striking similarity to HLH and has been associated with high IL-18.96 Features of HLH should be considered in all serious infections, and infectious disease consultation may be appropriate in many/most HLH patients. Lymphoma and leukemia are common malignancies associated with HLH, particularly T-cell and NK-cell lymphomas or leukemias, diffuse large B-cell lymphoma, and Hodgkin lymphoma.97 Solid tumors can also trigger HLH. It is particularly important to realize that HLH is due to an underlying malignancy in >50% of adult cases.98,99 Aggressive evaluations should be performed to evaluate for malignancy in adult patients with HLH. HLH can also occur during the course of chemotherapy and is often associated with an infection.97 Treatment with chimeric antigen receptor–modified T cells or bispecific T-cell–engaging antibodies can be associated with a cytokine release syndrome that mimics HLH.100,101
Diagnostic workup of patients with HLH
Once a diagnosis of HLH is suspected, several simultaneous assessments should begin (Figure 1). Laboratory and imaging studies should be performed to gather supportive evidence of a diagnosis of the syndrome of HLH and assess which organ systems are involved and the severity of involvement. Evaluations for infections and malignancies should be performed in all patients, including laboratory studies, bone marrow evaluation, general imaging, and biopsy of any suspicious findings. In children with suspicious clinical presentations, positron emission tomography imaging should be considered to evaluate for occult lymphoma. Aggressive oncologic and infectious evaluations are especially warranted in adults given that the majority of adult HLH is malignancy associated, followed by infection.98,99 Genetic mutations in genes regulating cytotoxicity are more rare in adults than children,102,103 and adult providers are referred to recent reviews for further discussion.104,105
In children and young adults, rapid protein/functional screening tests for genetic forms of HLH should be sent due to the delay in genetic testing results. As the turn-around time for genetic testing continues to decrease, genetic testing may become the first/only test. The likelihood of an underlying genetic diagnosis is highest in infants. At one large center, 61% of patients <1 year of age were found to have a genetic HLH disease compared with only 7% of patients aged 12 to 18.79 Ultimately, genetic diagnostic testing using next-generation sequencing panels or whole-exome sequencing is usually pursued, though targeted sequencing can still be performed when indicated (family history of a specific genetic defect, or a positive screening test result highly suggestive of a single likely gene [ie, absent perforin]). The differential diagnosis should be kept broad in patients for whom screening test results return normal, and in such cases, the laboratory evaluations should be expanded to look for other PIDs and metabolic diseases and reevaluate for occult infections and malignancies. The likelihood of an underlying genetic diagnosis in older adults is much lower than in pediatric patients, and the authors recommend consultation with an adult HLH specialist for recommendations regarding diagnostic evaluations in patients >20 to 30 years of age. However, as the cost of sequencing continues to decrease, one could make an argument for sequencing for all patients.
Rapid screening tests for genetic HLH
Several rapid screening tests are available to quickly screen for some of the genetic causes of HLH (Figure 1). Quantitative assessment of intracellular perforin, SAP, and XIAP protein is available clinically using flow cytometric analysis of peripheral blood cells and offers >80% diagnostic sensitivity.23,106 There are also adjunctive testing options such as measurement of invariant NKT cells (absent in SAP deficiency) or interrogation of NOD2 signaling (disrupted in XIAP deficiency). Abnormal cytotoxic granule exocytosis can be assessed using the CD107a (also known as LAMP1) degranulation assay. This assay measures surface CD107a on NK cells following a stimulus that induces degranulation. CD107a is found within the membranes of cytotoxic granules, but there is very little expressed on the surface of resting NK cells. As cells degranulate, CD107a is transiently expressed on the surface of the cell, and the increase can be measured by flow cytometry. The NK cell CD107a assay offers >90% sensitivity and, unlike the NK killing assay, does not appear to be greatly affected by steroid use.23,107 Combining perforin protein expression and CD107a mobilization to screen for familial HLH has better diagnostic accuracy than traditional chromium release NK-cell function testing.23
Treatment of HLH
Treatment of HLH should begin as soon as the syndrome has been recognized, with the caveat that steroids or chemotherapy should not begin until after evaluating for malignancy (with bone marrow, lymph node, tissue biopsy, etc.) so as not to interfere with diagnosis.97,108 Assessment for hemophagocytosis and staging for CNS involvement should not delay therapy.
The mainstays of HLH treatment consist of immunosuppressive and chemotherapeutic drugs and biologics that aim to dampen the cytokine storm and eliminate activated T-cell and macrophage populations. A commonly used treatment approach consists of dexamethasone and etoposide based on the experiences of the Histiocyte Society HLH-1994 and HLH-2004 studies. Patients with CNS involvement received additional targeted intrathecal treatment with methotrexate and steroid therapy. Many patients are still treated with this type of approach, and the Histiocyte Society recently published formal recommendations for the use of etoposide-based treatment.109 Approximately half of patients can be expected to achieve a complete response with steroid and etoposide treatment.9,11 A chemotherapy-sparing approach consisting of steroids, anti-thymocyte globulin, and cyclosporin A has been used more often in France, with a >70% remission rate.110
There are currently several agents in clinical trials or in the planning stages. As the array of possible choices for HLH treatment increases, we expect an increasingly individualized approach for patients based on their underlying etiology, disease severity, and pharmacogenomics. However, the lack of prospective study results makes formation of individualized plans difficult at this time. There is some experience with alemtuzumab,111,112 and a clinical trial is ongoing at the time of writing (NCT02472054). Emapalumab, a monoclonal antibody directed against IFN-γ, was recently approved by the Food and Drug Administration for HLH that is refractory, recurrent, or progressive or for patients who cannot tolerate conventional treatment,113 but clinical trial results have not been published yet (NCT01818492). A few case reports demonstrated efficacy of emapalumab in NLRC4 and other causes of MAS, and a trial of IFN-γ blockade in MAS is ongoing (NCT03311854).
Anakinra (recombinant IL-1 receptor antagonist) was recently found to be beneficial in a retrospective case series of secondary HLH, particularly when given early and to patients with underlying rheumatic disease.114 Agents targeting IL-1, tumor necrosis factor, and IL-6 have been reported in case reports and case series, especially for MAS, but no large studies have been performed. Ruxolitinib is a Janus kinase inhibitor that inhibits the signaling of several cytokines, including IFN-γ, and may hold promise for treatment of HLH based on murine efficacy data, a few human case reports, and a study of refractory/relapsed HLH.115-119 Three clinical trials were open at the time of writing (NCT03795909, NCT02400463, and NCT04120090). Anti-IL-18 directed therapy with a recombinant human IL-18–binding protein (tadekinig alfa) may be a good option for patients with diseases that are primarily driven by inflammasome activation and high IL-18 levels, such as patients with NLRC472 mutations or XIAP deficiency, and a clinical trial was ongoing at the time of writing (NCT03113760). Data regarding salvage therapy of patients with refractory HLH are sparse. Agents that have been used in the salvage setting include ruxolitinib, emapalumab, anakinra, alemtuzumab, antithymocyte globulin, and a liposomal doxorubicin + etoposide + dexamethasone regimen, among others.119-121
Treatment of HLH should be coupled with prompt treatment of any identified underlying trigger. Rituximab can be helpful in the treatment of EBV-HLH.122-125 Most patients should also receive aggressive antimicrobial prophylaxis directed against Pneumocystis jirovecii, general fungal organisms, and also viruses depending on previous exposures. IV immunoglobulin replacement is generally warranted. Patients with HLH can be critically ill and often need intensive care support in the early phase of diagnosis and treatment. Blood product support and correction of coagulopathy may be needed. Some centers use IV immunoglobulin, therapeutic plasma exchange, anakinra (recombinant IL-1 receptor antagonist), and/or corticosteroids to temper inflammation while workup is ongoing, although data supporting these practices are lacking. Patients with organ failures may require organ-specific interventions, and patients with CNS disease may need treatment of seizures or therapies for specific neurologic deficits. Allogeneic HCT is indicated in many pediatric patients with genetic HLH if a suitable donor is available, including patients with pathologic variants in PRF1, UNC13D, STX11, STXBP2, RAB27A, LYST, and SH2D1A and some patients with XIAP deficiency. Patients with HLH suffer from an unusually high rate of toxicities and mortality with fully myeloablative conditioning regimens,126 which is likely related to the underlying inflammatory state. Patients with XIAP deficiency are likely more susceptible to complications of graft-versus-host disease.127,128 Reduced intensity regimens are generally recommended, as they are associated with better survival, though they can be complicated by high rates of mixed chimerism and graft failure.129 Experience with reduced-toxicity conditioning approaches is growing. To avoid delays in proceeding to HCT, HLA typing and initiation of the stem cell donor search should be done as soon as it is suspected that a patient has a genetic form of HLH.
Concluding remarks
Pediatric HLH remains a challenge, but significant advances have been made in the last 20 years. We now know the genetic cause of HLH in the majority of patients with primary HLH, and whole-exome sequencing and panel-based testing has led to a broadened appreciation of immunologic and metabolic diseases that can be complicated by HLH other than the classically associated diseases. Advances in rapid screening diagnostics makes it possible to quickly evaluate patients for many inherited diseases, and newer biomarkers are helping to shed light on the driving physiologic processes in individual patients. Novel targeted treatment agents are being developed.
Nevertheless, several great challenges remain: (1) mobilizing HLH syndrome recognition and improving access to specific testing to minimize deadly delays in diagnosis/treatment; (2) continuing to develop clinical trials to determine which novel agents/combinations will result in optimal outcomes for patients with different, and sometimes multiple, underlying mechanisms of disease (eg, cytotoxicity defects, IL-18/inflammasomopathies, and metabolic disorders); and (3) directly comparing different treatment approaches for similar patients (eg, etoposide vs ruxolitinib, alemtuzumab, and emapalumab). Meeting these challenges will require cooperative support among investigators, national and international societies, and industry to systematically identify patients and complete well-designed clinical trials. With such efforts, the next 10 years promises to bring further improved outcomes for patients. Transplant approaches also continue to improve, but again, there is a great need to perform systematic trials designed to determine an optimal conditioning intensity which is both well-tolerated and allows sustained donor engraftment. There is meaningful work to continue in the HLH field, and unremitting efforts that are underway will continue to improve patient outcomes.
Authorship
Contribution: R.A.M. and S.W.C. wrote the manuscript.
Conflict-of-interest disclosure: S.W.C. has been a consultant for AB2Bio, Ltd. and received honoraria from Novartis, Inc. R.A.M. declares no competing financial interests.
Correspondence: Rebecca A. Marsh, Division of Bone Marrow Transplantation and Immune Deficiency, University of Cincinnati, Cincinnati Children’s Hospital Medical Center, 3333 Burnet Ave, Cincinnati, OH 45229; e-mail: rebecca.marsh@cchmc.org; and Scott W. Canna, Division of Rheumatology, University of Pittsburgh, UPMC Children's Hospital of Pittsburgh, Pittsburgh, PA; e-mail: scott.canna@chp.edu.